Charge Analysis
- When we solve the Schrödinger equation the energy is not the only output, we also obtain a wavefunction (Φ). The square of the wavefunction gives the electron density which can then be partitioned up and assigned "back" to each atom. The result is that we can determine a partial atomic charge due to the redistribution of electron density due to bonding within the molecule. This process can be quite technical but the results are very easy to understand.
- Open your final optimised structure *.chk file (without intermediate geometries!)
- Go to the results tab and select "charge distribution". A new window will appear under the type select NBO. You must select the NBO charge distribution not the default Mulliken (which is a poor method for computing charge). Then check the tick boxes as shown in the image to colour the atoms and to have the numerical charges present.
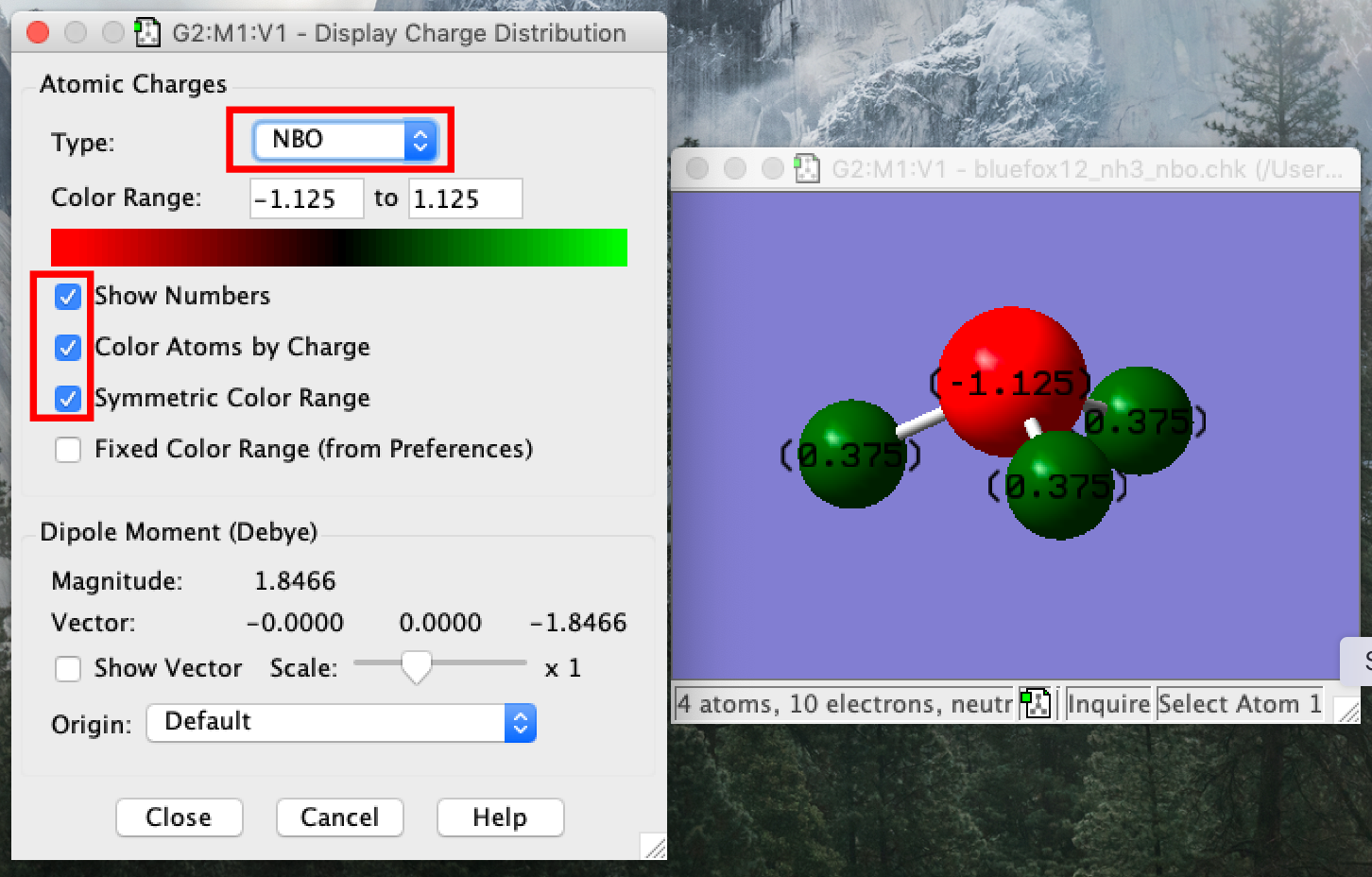
- Notice that gaussview has assigned a "Colour range", if you compare molecules, you must be careful to set manually set the range to the same value for both moleucles. Gaussview looks at your molecule and simply sets the range to the largest absolute values (and as we have the symmetric range box ticked both positive and negative ranges are set this way)
Now you know how to generate the charge distributions for a molecule!
- When you are ready close (just!) the charge distribution window move onto the next step
