
Carrying out a "Scan"
Introduction
Now you are going to form an IL ion-pair and then study the motion a H-atom (proton) could make between the ions, on one side the system is composed of neutral molecules, on the other it forms an ion-pair. Both H-bond!

We are going to take an optimised structure and "fix" this conformation, then we are going to move the H-bond atom along the N---Cl coordinate path and calculate the energy at each step to generate a "potential energy surface" PES.
Calculations
- important to reduce the time for calculations to run we will use the 3-21G basis set, and NOT use opt=tight, pop=(full,nbo) int=ultrafine or scf=conver=9.
- Create the ion-pair Me3NH-Cl.
- start with a "methyl"
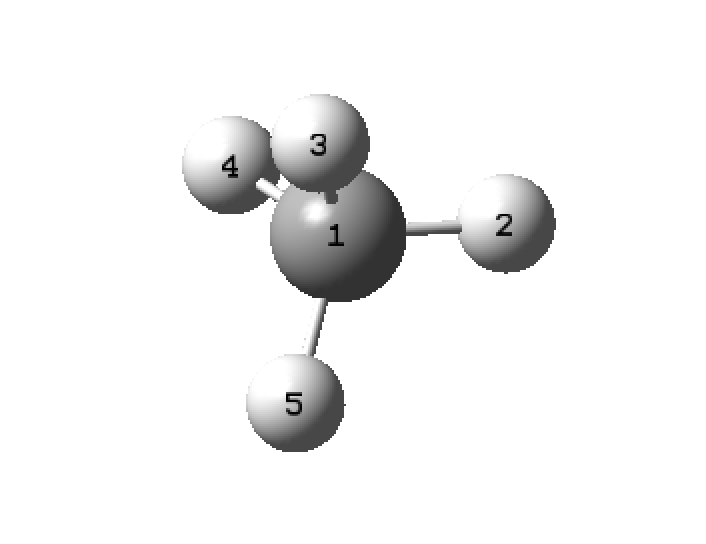
- select "view" and "labels" to give the atom numbers, carbon will be atom 1, align the molecule so H labeled 2 is lined up to the right
- replace the central C with N FIRST
- replace 3 of the H atoms with methyl groups
- Important! The N should now be atom 2, if it is not see below
- place the Cl along the N-H vector, make sure the N-H...Cl is linear by using the bond angle modification tool, fixing the N and H, and only allowing the Cl to move, make the angle 180 degrees.
- start with a "methyl"
- Optimise and confirm via a frequency analysis the ion-pair Me3NH-Cl. Don't worry about applying any symmetry. In your wiki (as should be obvious by now!) upload the log file, report the relevant convergence data and provide a jmol. In this case also report the N-H and N-Cl distances.
- Convergence
- an opt and freq job is essentially 2 calculations, the optimisation then frequencies
- when you look in your log file at the Item table, make sure to record the last (freq) and second-to-last (opt) tables. You will notice that one says the molecule is converged and the other says it is not!
- this is common for small molecules, poor basis-sets or flat PESs
- the optimiser updates the slope of the PES, where-as the frequency calculation exactly calculates the slope of the PES.
- if you are interested ask a demonstrator to explain why we can have poor convergence for a flat PES
- irrespective of the "Item" data for the freq calculations the most important point here is that the low frequencies are between ±10. If this is true and the forces are converged you are fine ... the distances can be "unconverged" up to an order of magnitude greater than the criteria.
- if the distances are too large get a demonstrator to check your calculation BEFORE proceeding
- we only record both Item tables if the freq shows any "unconverged" components
- it will be a good idea to have some discussion with the demonstrators about what convergence means and how we need to evaluate it ourselves and not slavishly follow the code output
- as in any real research project we report the results we have, even if they are not perfect
- now open your optimised structure (make sure it is showing the last step!!)
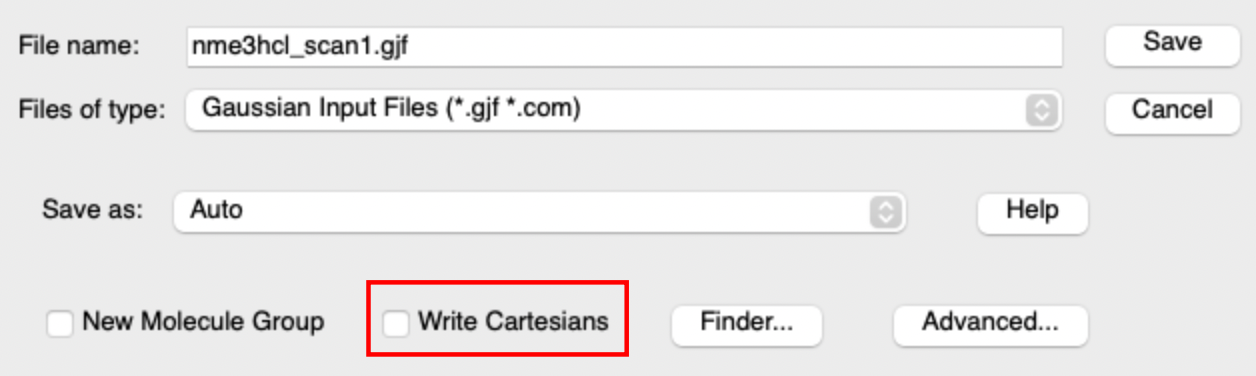
- save your file as a new *.gjf or *.com file, this is in-case you make a mistake and we need to start from your optimised structure again. When you save it make sure that the "write cartesians" box is NOT ticked.
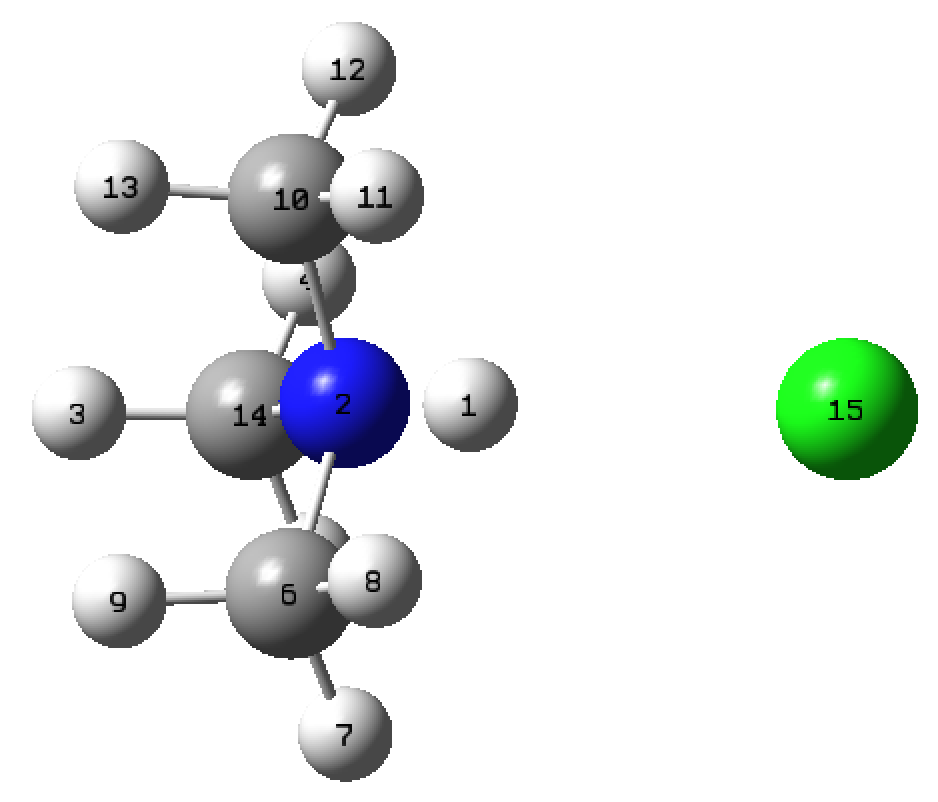
- use the bond tool to set the N to H distance to 0.8 Å and set the N to Cl distance to 3.2 Å
- check your atom numbering it should be N=2,H=1,Cl=15, if its not follow the switching labels procedure below
- save your file as a new *.gjf or *.com file, this is in-case you make a mistake and we need to start from your optimised structure again. When you save it make sure that the "write cartesians" box is NOT ticked.
- switching labels
- make sure atom numbering is visible
- in the Tools menue choose "Connection ..."
- you will see the window below pop up
- LEFT click on the atom no. 2, then RIGHT click on the N
- these two atom numbers will swap
- you can repeat until N=2 and H=1
- MAC users, this will not work unless you have a 3 button mouse, the mac mouse is unable to reproduce this action
- now goto the tools tab and select the atom list and select the "Z" and "O" options if they are not already active. Z stands for Z-matrix and make sure the N-H bond is 0.8 (first red arrow). O stands for opt and make sure this has Rxn/Scan (second red arrow).
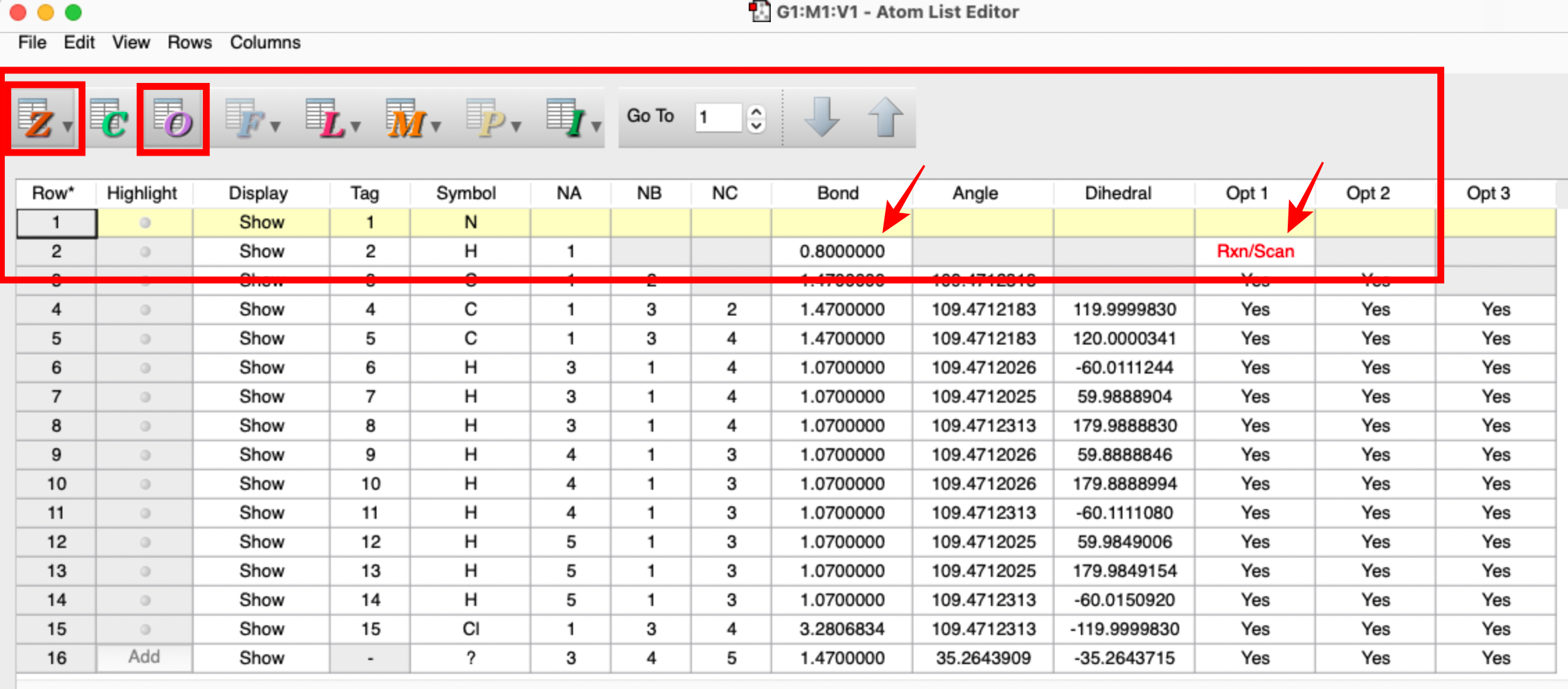
- then go to calculation setup and in the Job-Type tab select Scan rather than Opt+Freq and choose Rigid scan. Make sure the number of steps is 13, you start at 0.8 and the step size is 0.1.
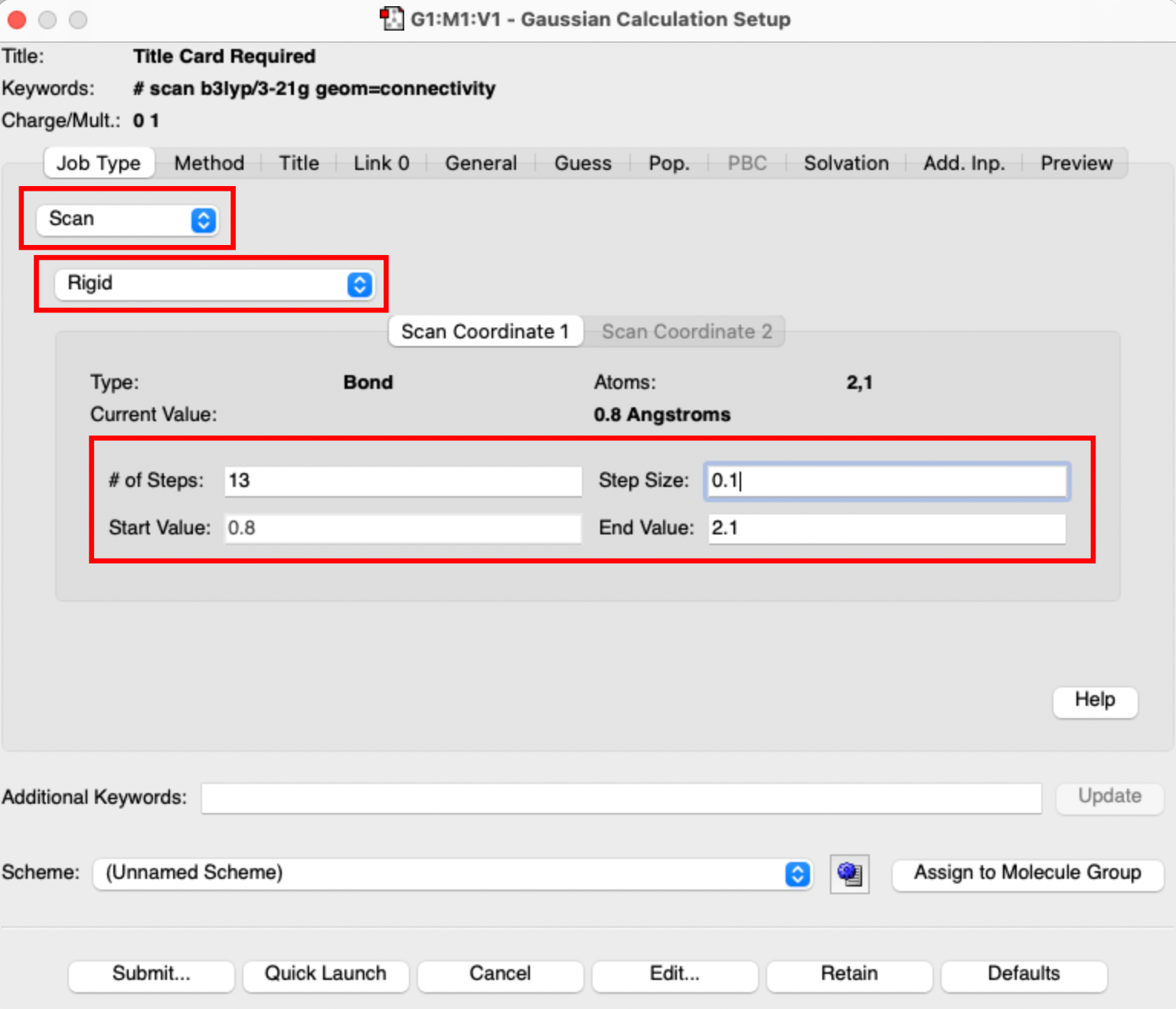
- submit this job to run under a new name, I called mine PH_me3nhcl_rigid_scan.gjf
- when the job finishes, open the log file. You should have 14 steps and be able to open the scan of the potential energy surface (Results then Scan). Click through the various points noticing how the position of the H-atom changes. The energy is given in atomic units. A graph like this is ok for checking your results and a "workbook", but it will never be suitable for a published report.
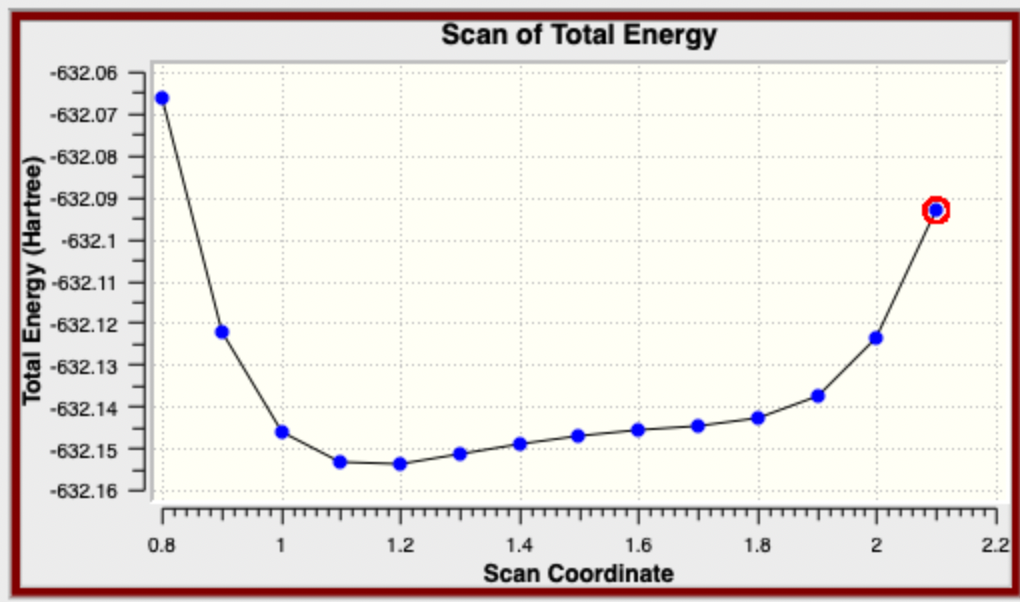
- you can right-click in the body of the graph and download the numerical data, this is what you will use to produce a graph. We report PES data relative to the lowest energy, and in kj/mol so you will need to manipulate the data a little before drawing the graph. Make sure to include a title and axis labels with units. You can see mine below.
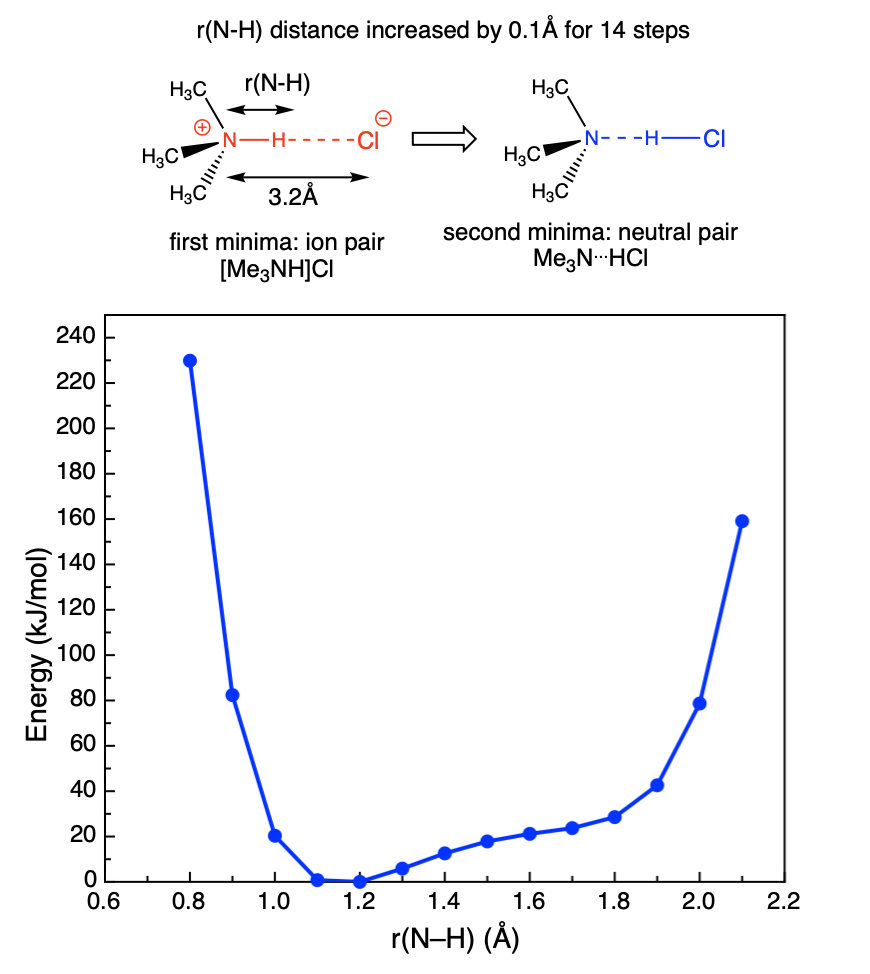
- In your wiki use text or a diagram to explain the scanned coordinate, and include both a snap-shot of the raw data and a report quality graph.
- this graph gives us important information, it shows us that when the N-Cl distance is set at 3.2Å a broad minima occurs, the most stable state is an ion-pair Me3NH+ --- Cl- and as the proton is pushed over to the Cl forming a neutral pair Me3N --- HCl the energy goes up, and no stable minima forms, rather there is a "shelf" in the PES. The ion-pair forms a doubly ionic H-bond between Me3NH+ and Cl-, the neutral-pair forms a normal H-bond between Me3N and HCl.
- now you know how to carry out a scan of a single coordinate (keeping the rest of the molecule fixed). Normally we would allow the rest of the molecule to "relax" at each point, this is called a relaxed scan, however this takes much more time!
- in the next section you will be carrying out a fixed scan of another ionic liquid pair!