Animating the vibrations
Now we are ready to look at the output from the frequency, or vibrational analysis. We will be animating the vibrations and looking at the computed IR spectrum.
- open your optimised structure (without intermediate geometries!)
- From the main menu along the top of the screen (in gaussview) choose "Results" and then choose "Vibrations":
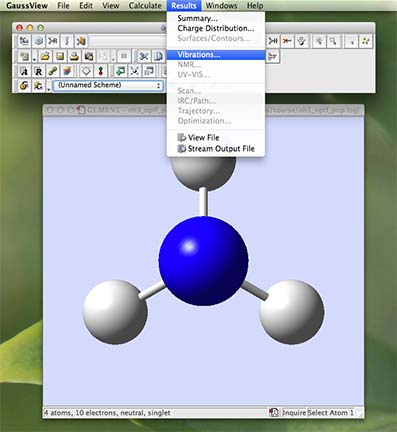
- a new window will open called "Display Vibrations":
- arrange your windows so that you can see both the molecule window and the vibration window
- in the molecule window rotate your molecule so it is not completely in the plane of the screen
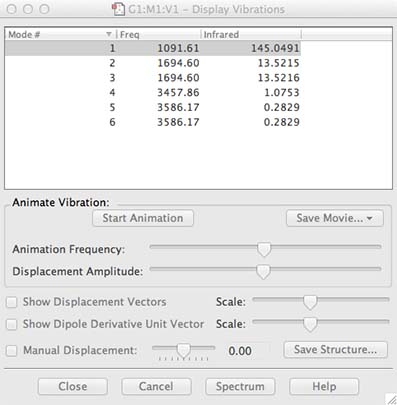
- in the vibration window highlight the top vibration, then check the "Show Displacement Vectors" box, and finally click on the "Start" button, the molecule should start vibrating!
- look at all the vibrations by highlighting them one after another down the list
- All of these vibrations are active at zero kelvin! They represent the zero-point energy of the molecule, this is a purely quantum mechanical property of molecules.
- If you have any vibrations with a negative number under the frequency heading this means your molecule is not fully optimised, go back and repeat the optimisation.
- you should have 6 vibrations, with frequencies similar (but not necessarily identical) to my example. The "mode" number is meaningless it is just to list the frequencies. The number in the IR column identifies the intensity of each vibration.
- the code is not "smart" it doesn't know to how many significant figures to report the results, so it just includes as many as it has. You as a human have to know how accurate the method and basis set you are employing are. In this case for B3LYP and 6-31G(d,p) both the wavenumbers and intensity are only accurate to integer values. It is very important that you report your numbers to the correct level of accuracy. The level you report the numbers to tells the reader how accurate they are!
- now look inside your log file, here is part of my log file:
Low frequencies --- -11.5222 -11.4865 -0.0030 0.0245 0.1415 25.6160
Low frequencies --- 1089.6618 1694.1735 1694.1738
Diagonal vibrational polarizability:
0.1276753 0.1276764 3.2989135
Harmonic frequencies (cm**-1), IR intensities (KM/Mole), Raman scattering
activities (A**4/AMU), depolarization ratios for plane and unpolarized
incident light, reduced masses (AMU), force constants (mDyne/A),
and normal coordinates:
1 2
A1 E
Frequencies -- 1089.6618 1694.1735
Red. masses -- 1.1800 1.0644
Frc consts -- 0.8255 1.8001
IR Inten -- 145.4481 13.5571
Atom AN X Y Z X Y Z
1 7 0.00 0.00 0.12 -0.07 0.00 0.00
2 1 0.00 -0.21 -0.53 0.76 0.00 0.00
3 1 0.18 0.11 -0.53 0.08 -0.39 0.22
4 1 -0.18 0.11 -0.53 0.08 0.39 -0.22