Optimising a Molecule of NH3
- if it is not already the front window, bring the window with your NH3 molecule to the front.
- now we are ready to set up the commands that tell the program how to do the calculation, we tell it
- the method: B3LYP
- the basis set: 6-31G(d,p)
- and what type of calculation to do: OPTF (optimise and frequency analysis)
- The method B3LYP determines the type of approximations that are made in solving the Schrödinger equation.
- The basis set determines the accuracy, 6-31G(d,p) has a medium level accuracy, using this basis set means the calculations should be quick.
- In carrying out an optimization we are determining the optimum position of the nuclei for a given electronic configuration, this is the first step for any quantum chemical calculation.
- Depending on the gaussview set-up some of these options may be pre-set for you or you may have to select them yourself, you can also explore the buttons and options but please come back to the defaults
- We are also going to add a few extra commands as well to allow us to investigate the vibrational modes and electronic structure of the optimised molecule.
- From the main menu along the top of the screen (in gaussview) choose "Calculation" and then choose "Gaussian Calculation Setup...":
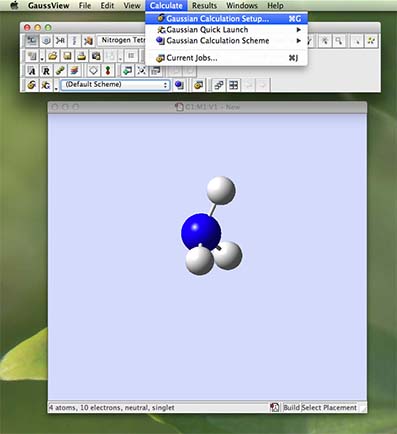
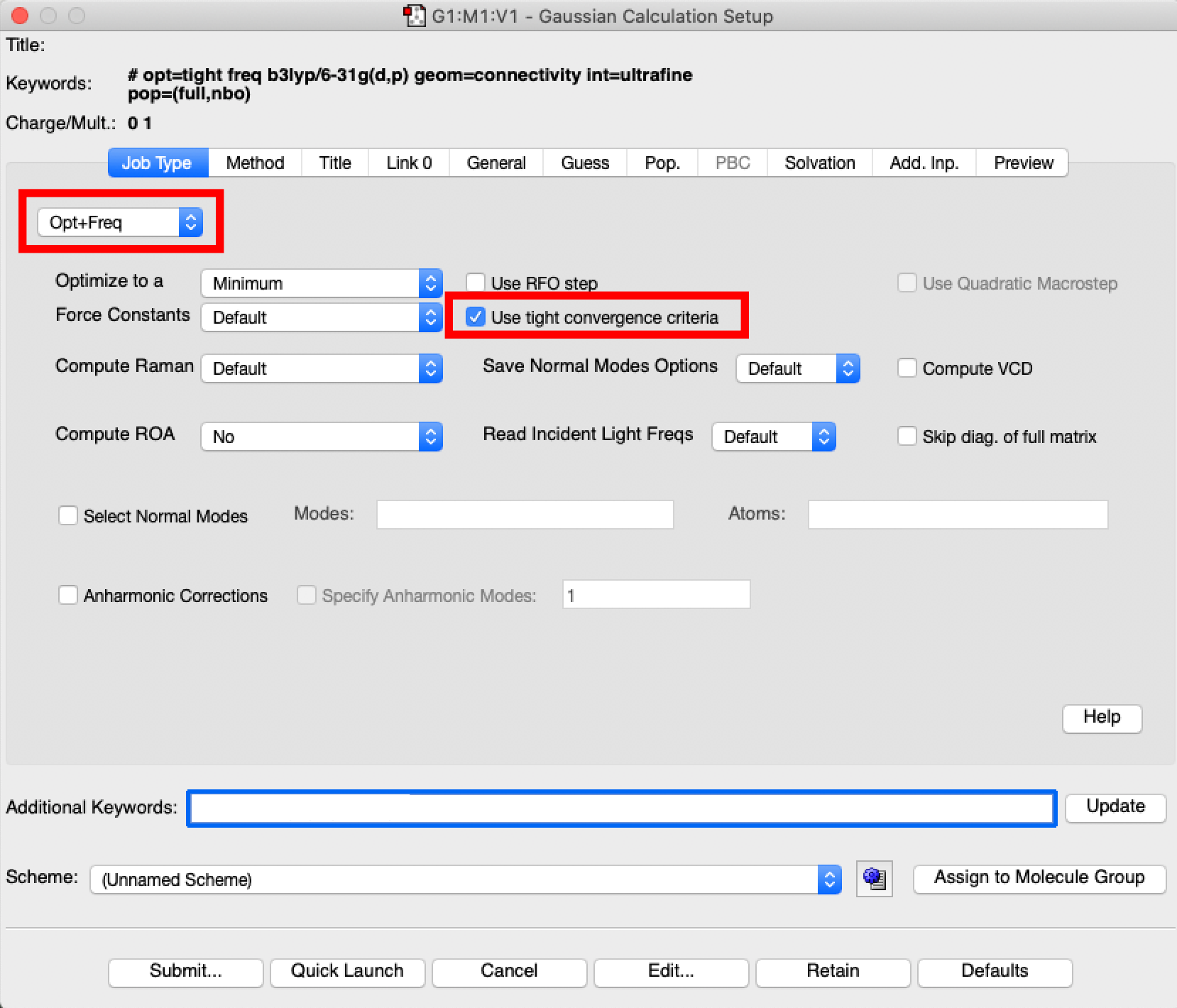
- a new palette will appear the calculation palette. It opens with the "job type" tab open. If the default is not set to "opt+freq" use the pull down menu to choose "opt+freq". This command requests that the calculation carry out an optimisation and then vibrational analysis. In addition you need to tick the "use tight optimisation criteria box".
- next click on the "method" tab to open it, the default should be B3LYP for the method and 6-31G(d,p) for the basis set. There is also some other information in the "Additional Keywords" section this should say int=ultrafine OR integral=grid=ultrafine (these are just 2 ways of setting this option), this option relates to how accurately the electronic density is computed, we want it to be computed on an ultrafine integration grid.
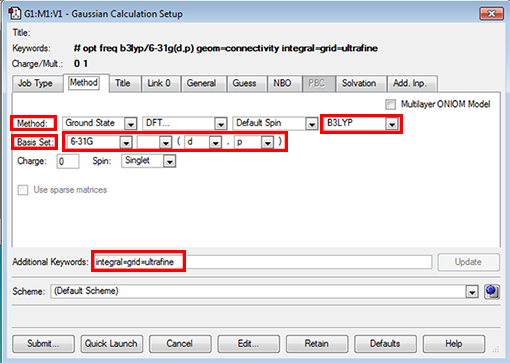
- If you don't see expected defaults then you can set the options yourself:
- use the pull down menu under "Hartree-Fock" to pick "DFT"
- find the pull-down menu "LSDA" and choose "B3LYP"
- in basis set choose 6-31G in the first button
- in basis set leave the 2nd button, but in the 3rd button choose "d" and in the 4th button choose "p"
- type into the "additional keywords" section: integral=grid=ultrafine
- We want to add two more commands, the first NBO will ask the code to run a population analysis so the atomic charges are calculated and the second pop=full to record all the MOs so we can visualise them (the main objective of this lab). Type into the additional keywords section "pop=(full,nbo)" as shown below:
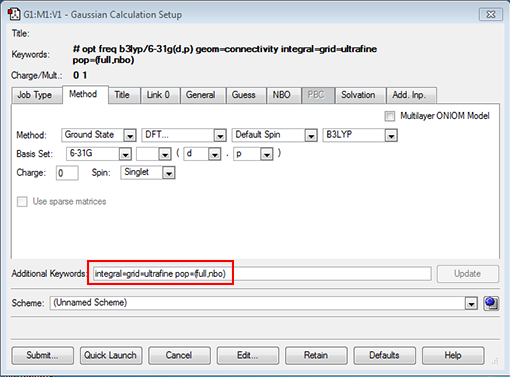
- We should also add a title for your job, so under the "title" tab type in something descriptive about the job I used "nh3 optimisation".
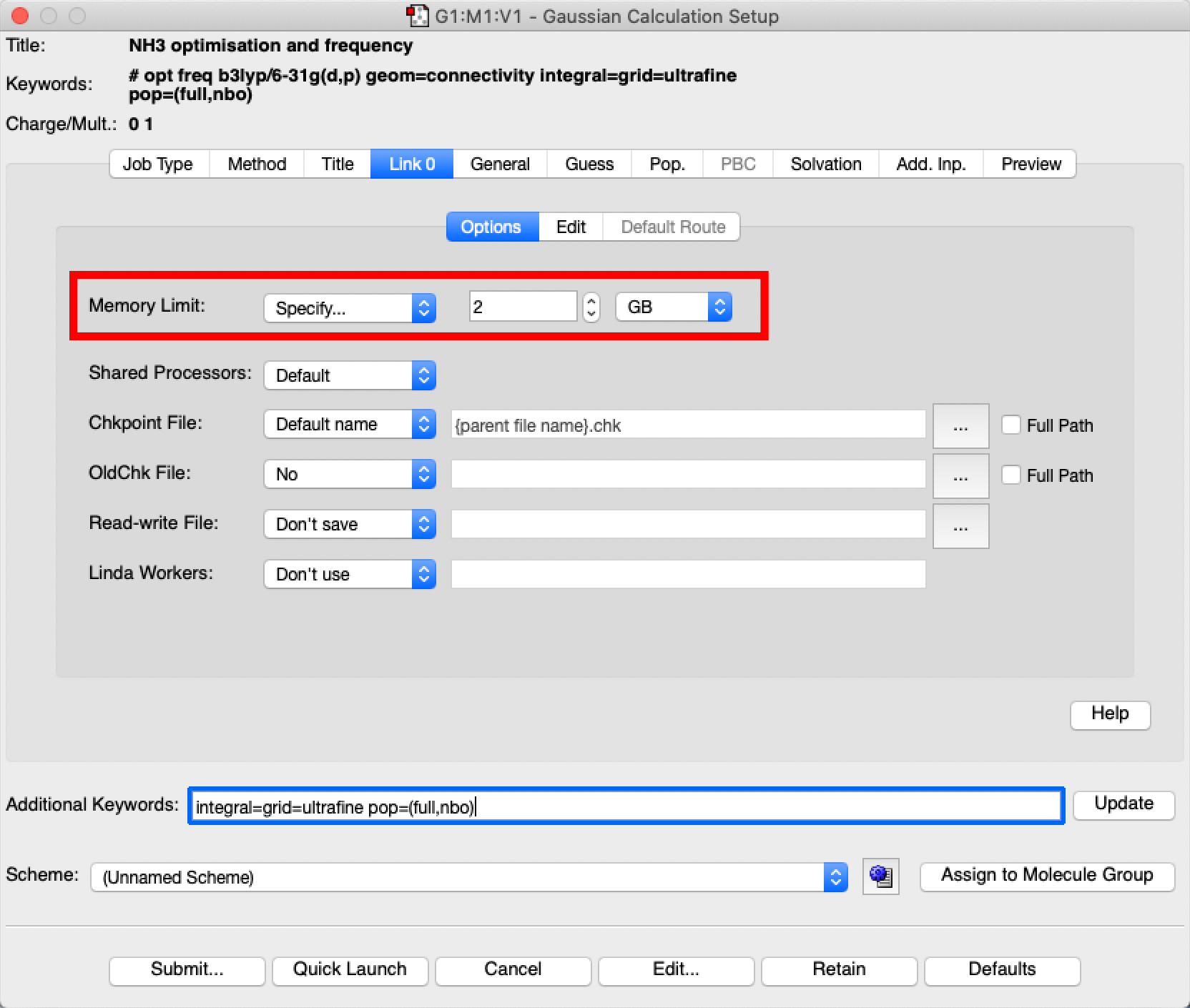
- Last and very important we need to set the amount of memory your job is allowed to use. Select "specify" on the pulldown menu and set this to 2 GB as shown below:
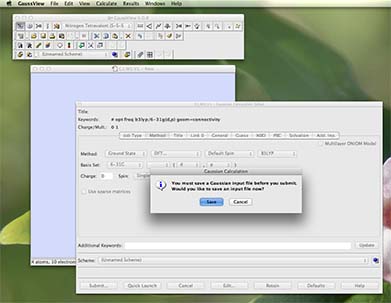
- Now you are ready to go! Press submit (button is in the bottom left of the palette)
- a new window will pop-up and telling you that you must save the file first, press "save":
- a new window will then pop-up and ask you for a file name.
- first navigate to the D: drive where we will save the file, or you could save to a flash drive. Do NOT save onto the C:drive
- linux does not like spaces in filenames NO SPACES in filenames use an underline "_" where you would use a space
- include your initials in your filename
- until you are experienced, remembering what is in every file can be difficult, give your file a short but descriptive name
- for example, my initials are PH, this is a nh3 molecule and we are doing an optimisation, so I would name my file
PH_nh3_opt.gjf
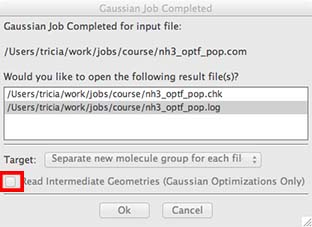
You should also select the "Read Intermediate Geometries" box as well.