Introduction to Moleuclar Orbitals
- You have already started learning about Molecular Orbital (MO) theory, and you have a number of courses that cover different aspects of MO theory from the mathematics through to qualitative MO diagrams in the 2nd Year. MO theory is also used in organic steroelectronics and in reaction mechanisms, both for transition metal complexes and organic synthesis.
- To help you better understand the information your lecturers will be presenting we are going to have a look at some MOs now. We are going to focus on the MOs of N2.
- Open the *.chk file for your N2 optimisation/frequency file. Goto the "File" pulldown menu, choose "open", then in the File type: select *.chk, and select your N2 checkpoint file
- Then from the "Edit" pulldown menu, choose "MOs":
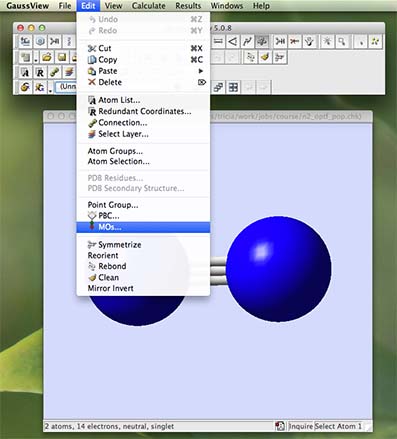
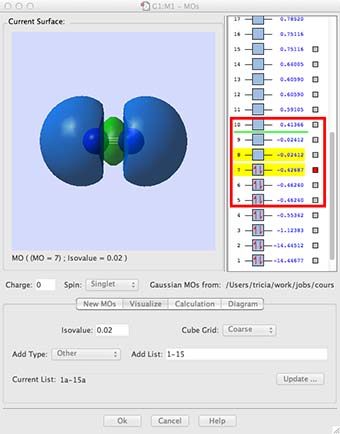
- A MO window should open. In the new window select the "Visualize" tab and then in the box "Add List" type "1-12", and click on Update. What will happen next is that gaussview will read information off the checkpoint file and covert this into the molecular orbitals 1-12 for you to visualise. Be patient, this requires quite a bit of number-crunching and may take upto 10min.
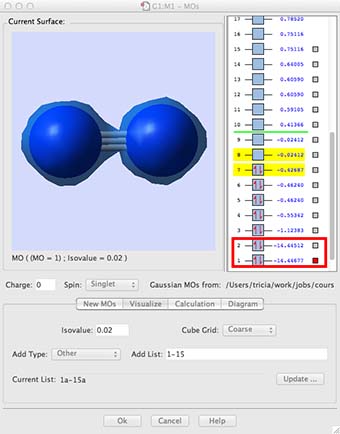
- While you wait, let me explain what the window is going to present (see below). On the right hand side are a list of numbers, these are the energy of the MOs in atomic units. In the body of the window the molecular orbitals are going to appear. These orbitals are the orbital solutions to the Schrödinger equation, in other words the Φ, one for each MO energy level.
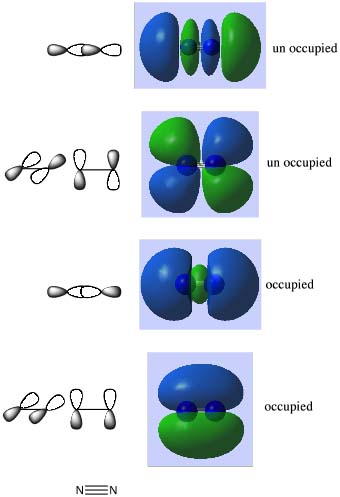
- You will know the electronic configuration of a nitrogen atom: 1s22s22p3. When the nitrogen atoms combine to form N2 this is actually the atomic orbitals (AOs) combining to form molecular orbitals (MOs)
- Click on the box for the lowest energy MO, the MO is the blue shaded region on the diagram above. (you may have different preferences set so it may give a mesh or an opaque surface, if you want to change the representation go into the "preferences" menu and select surfaces.) It is easy to see how this MO is a bonding combination of the two 1s core AOs on each N-atom. The next MO up in energy is the antibonding combination of the two 1s AOs. Notice how deep in energy these MOs are, -14.44 au much deeper than the MOs formed from the valence shell AOs. These AOs hardly overlap at all, they are held tightly to the respective nuclei, they are not very involved with chemical bonding.
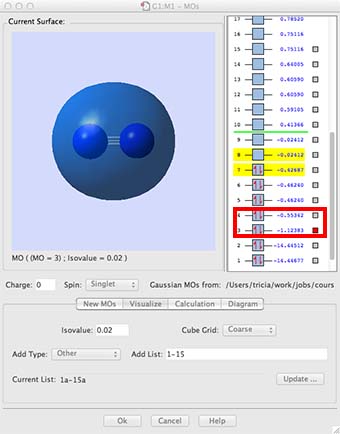
- Now look at MOs 3 and 4, this is the combination of the 2s valence AOs, bonding and antibonding. Notice how they overlap more strongly, the bonding overlap is so extensive that all we see is one extended surface. The energy is also much higher, now in the region of -0.5 to -1.12 au. The energy difference between the bonding and antibonding orbitals is much larger because they overlap better. These are valence orbitals and they are very involved in the chemical bonding.
- The next band of MOs are made up from the 2p AOs.
- There are two types of interactions, sigma along the bond and pi perpendicular to the bond, see the diagram below.
- It is easy to see how pAOs perpendicular to the bond sitting side by side form the low energy bonding MOs. And because there are two pAOs on each atom that are perpendicular to the bond, we get two MOs of exactly the same energy (-0.46 au), orbitals of the same energy are said to be degenerate. You might already know that this type of MO is called a pi MO.
- Above lies the MO formed from the bonding overlap of the along bond pAOs. There is a special reason called "mixing", which you will learn about a bit later in the year, which gives rise to the distorted shape of this MO. So remember these shapes (or just pop into the computer room and recalculate N2 when the time comes).
- The next two orbitals are again degenerate and these are the antibonding combination of the perpendicular pAOs.
- Then finally we have the antibonding MO from the along bond pAOs. This orbital is so high in energy it has positive energy, +0.41 au).
- One of the reasons N2 is so stable, is that only bonding pAO based MOs are occupied.
- You could also look at the higher energy MOs, however their shapes are not as reliable as those of the bonding MOs particularly the higher energy ones. When you get to your quantum mechanics course you will learn that this is because the SCF procedure only variationally optimises the occupied orbtials. So in terms of the mathematics the way we arrive at the un-occupied MOs is not as robust.
Now you are finished the "set" section of the lab, tidy your wiki up and ask for a rapid-feedback session from one of the demonstrators to ensure you are on the right track. After that it will be time for you to investigate your own choice of small molecule, there are some suggestions back on the main page for you to try.