
Understanding the Optimisation Process
Next you are going to optimise your NH3 molecule, but first I want to explain what the computer is doing for you and then we will set the job running.
Let us start by considering the energy E of a molecule. When we solve the Schrödinger equation (EΦ=HΦ) under the Born-Oppenheimer approximation we assume the nuclei are fixed, and we solve for the electrons (the wavefunction Φ) and energy (E). You will learn more about this in your "Chemical Bonding" course in 2nd year.
In fact, you are already familiar with this concept! The dissociation curve of a diatomic represents a potential energy curve E(R), that is, E dependent on R (the distance between the two atoms), see the figure below.
As the nuclei get close together, they repel each other (two positive charges repel) and the energy goes up. As the nuclei become stretched and the electrons no longer form a bond, the electron-nuclear attraction decreases, the energy goes up and the molecule dissociates.
Somewhere in the middle the electrons and nuclei are able to arrange themselves so that they are stable, we say that the system is in equilibrium. When something is in equilibrium we say there are no net forces on the atoms within a molecule (nothing pushing on them to make them want to move).

When carrying out calculations, the first thing we do is choose the position of the nuclei, this is the information you have provided in building your molecule. Rather than specify each position independently I'm going to define R as a collective coordinate which represents all the atomic positions.
We then solve the Schrödinger equation for the fixed nuclear positions, and obtain an energy E and wavefunction Φ which are dependent on the frozen position of the nuclei, we write this as: E(R) and Φ(R1). This process is called a self consistent field (SCF) calculation. During the SCF calculation we are finding the electronic wavefunction (Φ) that gives the lowest energy (for the fixed nuclei). This is also sometimes called a "single point" calculation.
If I move the nuclei slightly I will get a different energy E(R') because the interactions inside of H will be slightly different. For example the electrostatic interaction between the various nuclei will be different because the distance between them has changed.
The nuclear postitions we have choosen when building our molecle are unlikely to be the correct ones, and so we must optimise the structure to find the position of the nuclei that gives the lowest energy E(Ropt). That is we need to find the equilibrium or lowest energy geometry (represented by Ropt) as shown on the figure above.
First the nuclear positions (R1) are set and an SCF cycle is run to find the energy E(R1) and wavefuntion Φ(R1). Then we move the nuclei a small amount to positions represented by R2 and the SCF cycle gives us E(R2) and Φ(R2). Then we choose the positions, R1 or R2 that give us the lowest energy, thus if E(R2) < E(R1) we choose R2
Normally we need many more than 2 steps, and there are "tricks" for directing the search so that we can achieve a fully optimised structure very quickly.
Thus we compare the energies E(R2) and E(R1) if the new energy is lower we "move" in that direction. Then we start our search again from this new nuclear configuration. In this way we gradually move toward the lowest energy structure.
As the system moves there will be a sequence of different energies all dependent on the changing coordinates of the nuclei and electrons. We are traversing a Potential Energy Surface (PES), the potential energy surface E(R) for a diatomic has already been shown above. The process of optimization is shown in the figure below.
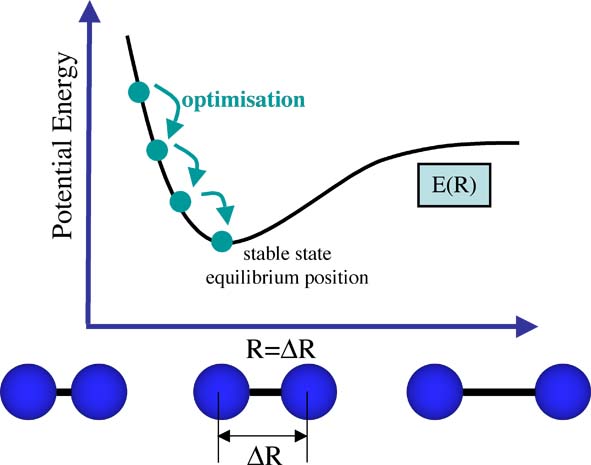
When the nuclei and electrons are in equilibrium, they experience no forces trying to shift them one way or another. When the nuclei and electrons are not in equilibrium their mutual interactions, ie nuclear-nuclear repulsion, nuclear-electron attraction, electron-electron interactions are not stable, these interactions cause forces to be exerted on the nuclei and electrons making them shift into better positions.
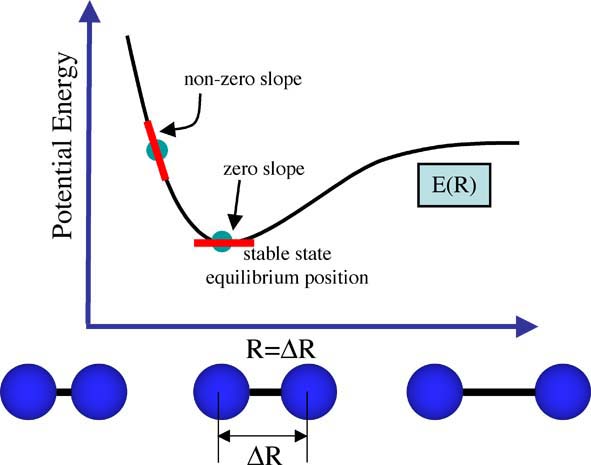
That is, the molecule will experience forces F(R) as long as a change in position (delta R) causes a change in energy, delta E(R). This equation is a first derivative, and we can relate it to the E(R) function, drawn above for the diatomic, as the slope of the graph at any point.
And at equilibrium (when we have zero forces) the slope of the graph is zero, see the figure below. This is also something you should know from your math courses. When the system is not in equilibrium the slope of the E(R) graph is not zero. Thus we now have a criteria for searching for the structure of a molecule, the structure we want is the one at which the first derivative of the PES is zero.
This is the process the computer carries out for us when we perform an optimisation
When you are ready move onto the next step