Understanding optimisation part b
What is happening when we carry out an optimisation?
Let us start by looking at the energy E. When we solve the Schrodinger equation under the Born-Oppenheimer approximation we assume the nuclei are fixed, and we solve for the electrons. The key point is that we obtain an energy E which is dependent on the frozen position of the nuclei. If I represent all the nuclear positions by a collective coordinate R, I get an energy E(R), an energy dependent on the frozen nuclear positions. All this means is, that if I move the nuclei slightly I will get a different energy E(R') because the interactions inside of H will be slightly different. For example the electrostatic interaction between the various nuclei will be different because the distance between them has changed.
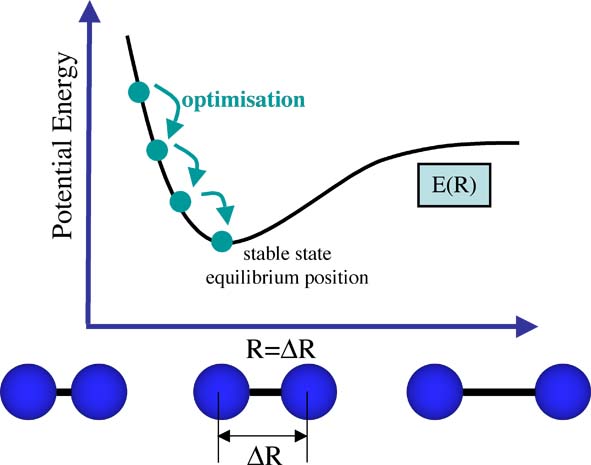
In fact, you are already familiar with this concept! The dissociation curve of a diatomic represents a potential energy curve E(R), that is, E dependent on R, see the figure below. In a diatomic there is only one "R" the distance between the two nuclei. As the nuclei get close together, they repel each other (two positive charges repel) and the energy goes up. As the nuclei become stretched and the electrons no longer form a bond, the electron-nuclear attraction decreases, the energy goes up and the molecule dissociates. Somewhere in the middle the electrons and nuclei are able to arrange themselves so that they are stable, we say that the system is in equilibrium. When something is in equilibrium we say there are no net forces on the molecule (nothing pushing on them to make them want to move).

When we optimise a structure we are looking for this stable point. That is we solve the Schrödinger equation for the nuclear positions and electrons and find the energy E(R) (again I've represented the collective coordinates by R). Then we move the atoms a little solve the Schrödinger equation again obtaining a new energy E(R') (I've represented the shifted collective coordinates by R'). Then we compare the energies and if the new energy is lower we "move" in that direction. Then we start our search again from this new nuclear configuration. In this way we gradually move toward the lowest energy structure. As the system moves there will be a sequence of different energies all dependent on the changing coordinates of the nuclei and electrons. We are traversing a Potential Energy Surface (PES), the potential energy surface E(R) for a diatomic has already been shown above. The process of optimization is shown in the figure below.
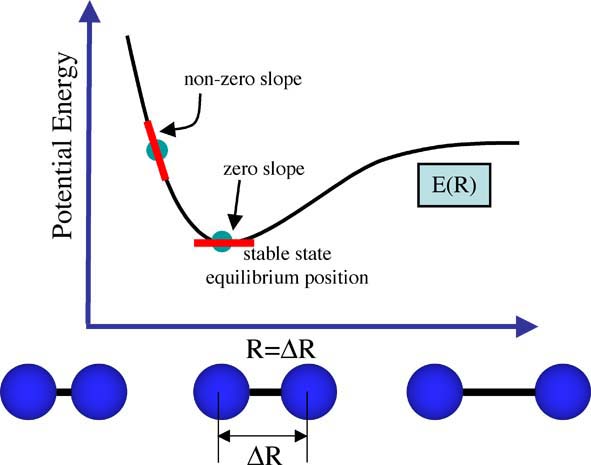
When the nuclei and electrons are in equilibrium, they experience no forces trying to shift them one way or another. When the nuclei and electrons are not in equilibrium their mutual interactions, ie nuclear-nuclear repulsion, nuclear-electron attraction, electron-electron interactions are not stable, these interactions cause forces to be exerted on the nuclei and electrons making them shift into better positions. That is, the molecule will experience forces F(R) as long as a change in position (delta R) causes a change in energy, delta E(R). This equation is a first derivative, and we can relate it to the E(R) function, drawn above for the diatomic, as the slope of the graph at any point. And at equilibrium (when we have zero forces) the slope of the graph is zero, see the figure below. This is also something you should know from your undergraduate maths courses. When the system is not in equilibrium the slope of the E(R) graph is not zero. Thus we now have a criteria for searching for the structure of a molecule, the structure we want is the one at which the first derivative of the PES is zero.
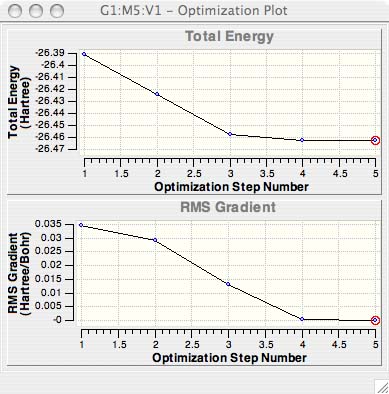
What information is given by the graphs in Gaussview?
The Total Energy curve, shows the program traversing the PES of BH3 and finding the minimum energy structure. The second graph is the RMS (Root Mean Square or average) Gradient, and shows the gradient going to zero as we approach the minimum.