
Using pseudo-potentials and larger basis sets
Important Carry out this calculation on the HPC. There are instructions on how to do this below.
Create a Molecule of GaBr3
- First, if you have any other "molecule" windows open, close them
- Then choose file -> new -> create mol group. A new blank molecule window should appear
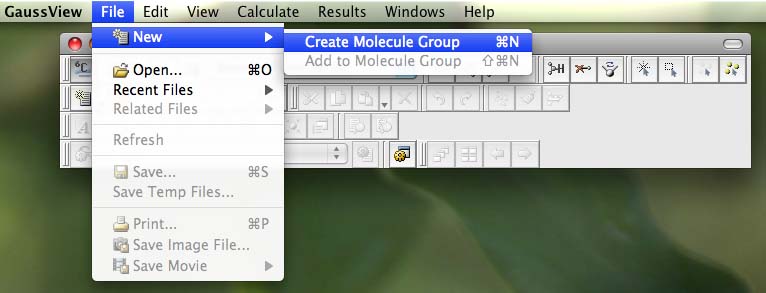
- on the controls palette click on the button that has a grey periodic table (or element fragment) on it:
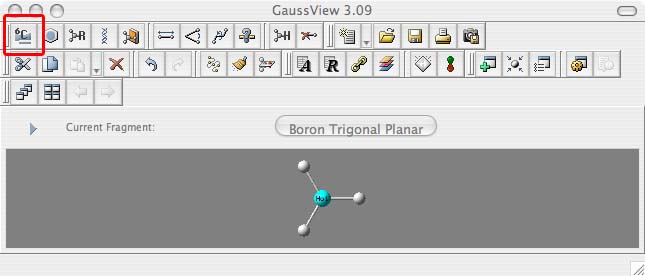
- when the periodic table comes up choose Ga for Gallium (1.), then when the options appear along the bottom of this window choose the trigonal planar fragment (2.):
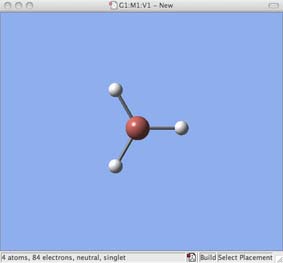
- now move the mouse to the molecule window and click once inside this window, a molecule of GaH3 will appear:
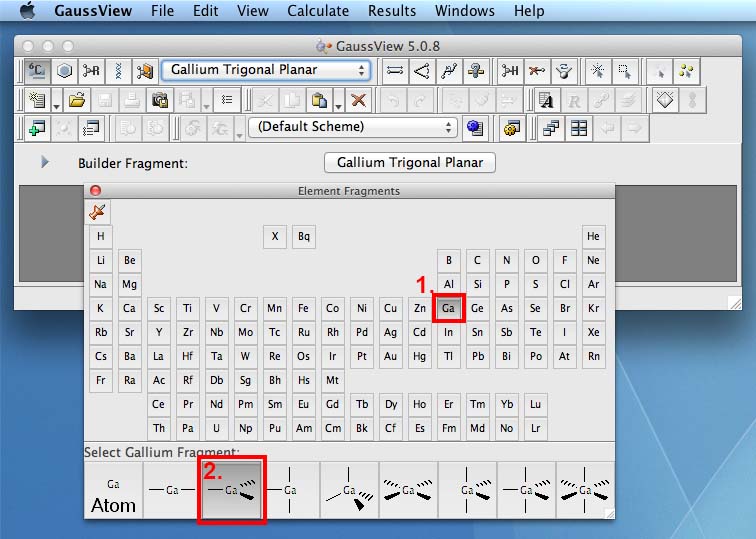
- now go back to the periodic table and choose Br for bromine (1.), then when the bonding options appear along the bottom of this window choose the atomic fragment (2.):
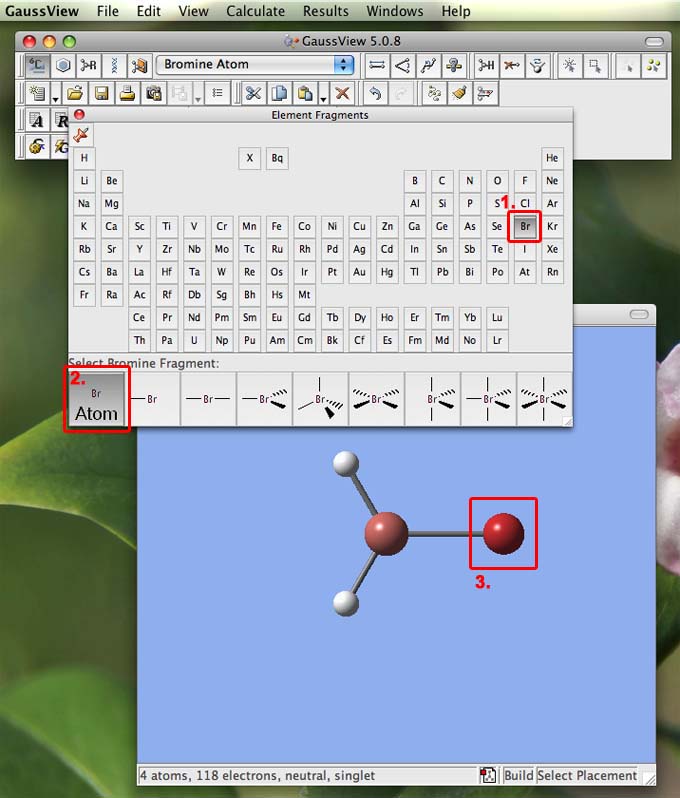
- then move the mouse to the molecule window and click ontop of one of the H atoms, it will turn into a Br atom (3.). Do the same for the other two Br atoms to make GaBr3. It is worth noting that this "trick" will work with functional groups as well as atoms.
- on the controls pallette click on the button with a question-mark in front of a ruler. This is the inquiry button which we will use later, however once in inquiry mode the other buttons deactivate and this will stop you accidentially making changes to your molecule.
Restricting the Symmetry of GaBr3
- we do not normally set the symmetry, this situation is a special case, however this is something it is very useful to know how to do
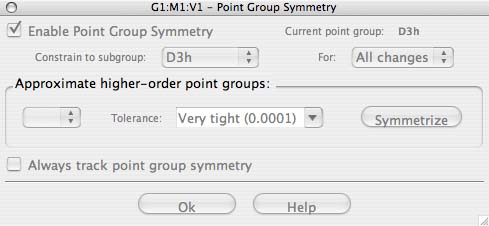
- to set the symmetry, goto the edit tab and click on the "point group", a new box will appear, click on the tick-box for "Enable point group symmetry, and choose to constrain the point group to D3h. Then increase the tollerance from the default to "Very tight (0.0001)" and then click on OK.
- if you don't set the tolerance to a high value there will be a problem with your vibrations and MOs later
Set up the calculation for GaBr3
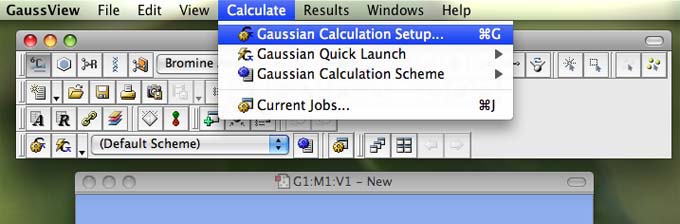
- From the main menu along the top of the screen (in gaussview) choose "Calculate" and then choose "Gaussian calculation setup":
- a new palette will appear the calculation palette. It opens with the "job type" tab open. Use the pull down menu under "energy" to choose "optimization" (ignore the other stuff that appears)
- then click on the "method" tab to open it, check that on the method line the second box says "DFT..." and the last box says "B3LYP". Click on the B3LYP pull-down menu and have a look and see what the other options are (don't change this from B3LYP!)
- then just below this where is says "basis set" use the pull-down menu to choose the LanL2DZ option, this uses a medium level basis set: D95V on first row atoms and Los Alamos ECP (ie pseudo potentials) on heavier elements. You only need to know that these are "medium" level you don't need to remember the names, the reason we don't use better basis sets and pseudo potentials is that the best ones are a bit more technical to implement and will make the calculations take much longer!
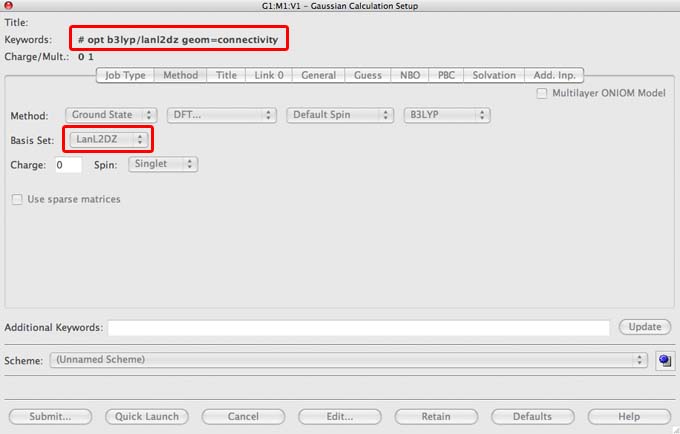
- check that the keywords in the red box in the image above are in your file, note that some of the lab computers are set up automatically to add as a default the command: "integral=grid=ultrafine" if you don't see this in your file then please add this text to the "additional commands" section. This command improves the integration of the electron density and provides a more accurate result.
- then click on the "title" tab to open it, and type "GaBr3 optimisation using pseudopotentials"
- Click on "Submit..." this won't actually submit the file, but will bring up the option to save the file. Save your file with a descriptive name, I called mine TH_GaBr3_pp_optimisation.com. Then do not submit the job, just save it.
- You are going to submit this job to the scan HPC service follow the instructions here.
Check that your optimisation of GaBr3 has been succesful
- Visualise the final log file in gaussview to ensure it "looks" right, none of the atoms have dissociated or strange bonding patterns have emerged.
- Check the "Summary" data is everything as it should be? Check the method, the basis-set, the symmetry and the gradient.
- Confirm that your optimisation has worked opening the .log file and checking the "Item" table, does each line say "YES".
- ONLY if your job has converged properly deposit the final optimised GaBr3 file in the chemical database "D-space". Instructions for adding a file to DSpace and including a link on your wiki.
Update your wiki
- Link to your completed B3LYP/LANL2DZ GaBr3 optimisation file on your wiki
- Include the "summary" table in your wiki
- Include the "Item" table in your wiki
- Include a Jmol image of your optimised geometry in your wiki
- use the inquiry button to determine the optimised Ga-Br bond distance and the optimisd Br-Ga-Br bond angle, put this information in the table we created earlier on your wiki
- Here is an example of my progressing wiki page.
When you are ready move onto the next step