
Using a mixture of basis-sets and psuedo-potentials
Important Carry out this calculation on the HPC.
Create a Molecule of BBr3
- when compunds contain both heavy atoms which require a pseudo-potential, and light atoms, which are treated more accurately with a full basis set we need to be able to mix psueod-potentials and basis sets.
- Set up a calculation for BBr3: Part1
- open your 6-31G(d,p) optimised log file for BH3
- open the periodic table and select Br
- then select "atom" Br
- now click on each of your H's in turn, they should turn into Br's with longer bonds
- then goto "gaussian set-up calculation"
- select "optimisation" under the "Job Type" tab
- add a descriptive title
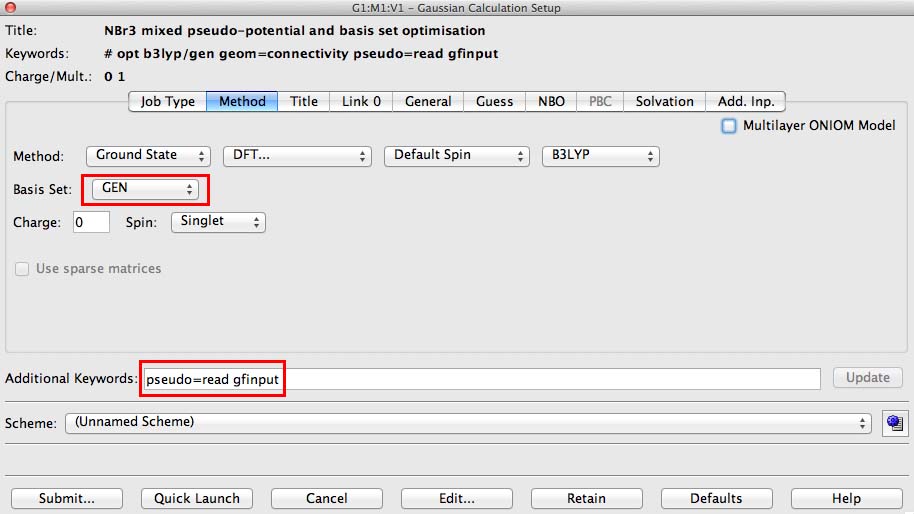
- under the methods tab on the basis button choose "GEN". This switches on the option to specify basis sets for each atom individually
- in the additional keywords section add pseudo=read gfinput. This switches on the option to specify pseudo-potentials for each atom individually and it turns on a printing option that is very useful if you make a mistake.
- note that some of the lab computers are set up automatically to add as a default the command: "integral=grid=ultrafine" if you don't see this in your file then please add this text to the "additional commands" section. This command improves the integration of the electron density and provides a more accurate result.
- then save your file, but do not submit it, ie press the submit option, save the file and then give no for the actual submission
- close the file
- now you need to add some information directly to the file (this is an advanced technique which gaussview does not support yet!)
- open the .com file you have just created
- go to the results tab and select view file, a window should open with your input file ready to edit.
- your file should look something like this:
%chk=bbr3_opt_mixed.chk # opt b3lyp/gen geom=connectivity pseudo=read gfinput integral=grid=ultrafine BBr3 mixed pseudo-potential and basis set optimisation 0 1 B 0.00000000 0.00000000 0.00000000 Br 0.00000000 2.02000000 0.00000000 Br 1.74937123 -1.01000016 0.00000000 Br -1.74937123 -1.01000016 0.00000000 1 2 1.0 3 1.0 4 1.0 2 3 4
- Now we need to specify the basis sets for all atoms and the pseudo-potential for the Br atoms. This is done after the coordinates and connectivity information. I have an example below for the BBr3 molecule where I want a full basis set on the B but PPs on the Br atoms:
%chk=bbr3_opt_mixed.chk # opt b3lyp/gen geom=connectivity pseudo=read gfinput integral=grid=ultrafine BBr3 mixed pseudo-potential and basis set optimisation 0 1 B 0.00000000 0.00000000 0.00000000 Br 0.00000000 2.02000000 0.00000000 Br 1.74937123 -1.01000016 0.00000000 Br -1.74937123 -1.01000016 0.00000000 1 2 1.0 3 1.0 4 1.0 2 3 4 B 0 6-31G(d,p) **** Br 0 LanL2DZ **** Br 0 LanL2DZ
the general format is the following:
coordinates
(blank line)
atomic symbols (space) 0 (zero)
normal basis set
**** (four stars)
atomic symbols (space) 0 (zero)
pseudo-potential basis set
**** (four stars)
(blank line)
atomic symbols (space) 0 (zero)
pseudo-potential
(blank line) important!
- add the extra information required into your file
- save your file in the text editor, close the text editor
- submit your job on the scan server. Instructions for submitting a file to the Scan servers.
Check that your optimisation of BBr3 has been succesful
- Visualise the final log file in gaussview to ensure it "looks" right, none of the atoms have dissociated or strange bonding patterns have emerged.
- Check the "Summary" data is everything as it should be? Check the method, the basis-set, the symmetry and the gradient.
- Confirm that your optimisation has worked opening the .log file and checking the "Item" table, does each line say "YES".
- ONLY if your job has converged properly deposit the final optimised BBr3 file in the chemical database "D-space". Instructions for adding a file to DSpace and including a link on your wiki.
Update your wiki
- Link to your completed B3LYP/6-31G(d,p)LANL2DZ BBr3 optimisation file on your wiki
- Include the "summary" table in your wiki
- Include the "Item" table in your wiki
- Include a Jmol image of your optimised geometry in your wiki
- use the inquiry button to determine the optimised B-Br bond distance and the optimisd Br-B-Br bond angle, put this information in the table we created earlier on your wiki
- Here is an example of my progressing wiki page.