we normally run a calculation at a low level to get the structure roughly right before using more expensive methods
now we will use your optimised geometry to start a new optimisation using a higher level basis set: 6-31G(d,p)
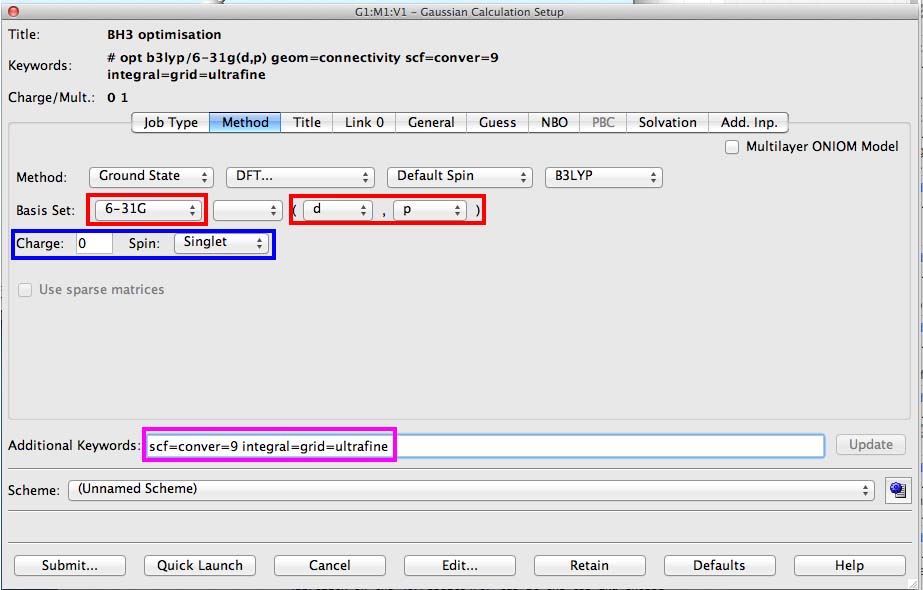
Set up for the 6-31G(d,p) calculation
open your 3-21G optimised log file
goto calculation setup and the method tab
on the basis set line change the basis set and then add the extra d and p funtions in the brackets, this is indicated in red on the diagram below
it is worth mentioning now the area shown in blue, this gives the charge and multiplicity of the molecule. BH3 is neutral charge=0. All the electrons are paired (S=0) and so the multiplicity (2S+1)=1. Gaussview quite often makes an incorrect "guess" for the multiplicity, so it is worth checking this in every calculation.
in the additional keywords section add "scf=conver=9", see the pink boxed area on the figure
some of the lab computers are set up automatically to add as a default the command: "integral=grid=ultrafine" if you don't see this then please add this text to the "additional commands" section. This command improves the integration of the electron density and provides a more accurate result
then submit the calculation
don't overwrite your existing file be sure to give the file a new name!
descriptive names are good, my first file I called bh3_opt_321g and this second file I called bh3_opt_631g_dp. It is important not to use special characters like brackets or commas in file-names, I use an underscore (which is allowed) to split up parts of a file name.
this file should run very quickly, mine only took two steps to complete.
Check your job has converged properly
click on the results tab at the top and choose "Summary". This will open a window that lists important information about your calculation.
check the gradient, has your job converged properly?
confirm that your optimisation has worked by looking at the "Item" table in the .log file
does each line say "YES"?
Total Energies VERY Important
reporting total energies
Look at the total energy for your 3-21G optimised structure (given in the summary table)
I obtained -26.46226433 au
you should have something similar, if not please see the demonstrator
Look at the total energy for your 6-31G(d,p) optimised structure
I obtained -26.61532363 au
again you should have something similar, if not please see the demonstrator
the numbers you obtain may not be exactly the same as mine, they may vary in the last decimal places. This variation is due to errors accumulating as the numerical calculations are carried out.
we only report total energies to allow others to check that their calculation, carried out with the same basis set and method, returns a similar energy to us, if it doesn't it means there is something wrong
the total energy for the 6-31G(d,p) calculation is lower (a more negative energy), so this computation is better, right? The energy difference is 0.1530593, this is a small number, right? NO to both!
the energy difference 0.1530593au, this is about 402 kJ/mol. This is a large amount of energy! (Compare this to the dissociation energy of a C-C bond which is about 350 kJ/mol, our molecule should be falling apart!)
The total energy for any calculation is highly dependent on the quality of the basis set. What this means is that we can NEVER compare the total energy of structures computed using different basis sets (or pseudo-potentials). The above comparison is meaningless!!
You can only compare energies for molecules computed with exactly the same method, AND the same number of atoms each computed with exactly the same basis-set (on every atom)
It is standard to report energies in kJ/mol up to 2dp (two decimal places)
Thus we need to report energies in au up to an accuracy of 0.01 kJ/mol, making the conversion this is 0.0000038 au, so report energies in au up to 7dp
see the web-page on "accuracy" for more information
Warning Because we have had too many people ignoring this extremely important concept in the past, anyone who:
attempts to compare total energies for molecules computed using different methods/basis sets will loose marks!
does not report their energies to the right number of significant figures will loose marks!
Update your wiki
Link to your completed 6-31G(d,p) BH3 optimisation file on your wiki
Include the "summary" table in your wiki
Include the "Item" table in your wiki
Include a Jmol image of your optimised geometry in your wiki
use the inquiry button to determine the optimised B-H bond distance and the optimisd H-B-H bond angle put this information in the table we created earlier on your wiki
When you are ready close the molecule window move onto the next step