
Using a mixture of basis-sets and psuedo-potentials
Create a Molecule of NI3
- each year I change the "heavy" molecule, so the images below may not match this year's molecule exactly.
- This year you will be looking at NI3, I is in the fourth row and has 53 electrons, with an electronic configuration of 1s22s22p63s23p63d104s24p54d105s25p5, at the level of quantum mechanics we are using, 53 electrons is a lot! In addition I does exhibit significant relativistic effects so we MUST use a PP.
- when compunds contain both heavy atoms which require a pseudo-potential, and light atoms, which are treated more accurately with a full basis set we need to be able to mix psueod-potentials and basis sets.
- Set up a calculation for NI3: Part1
- open your 6-31G(d,p) optimised log file for NH3
- open the periodic table and select I
- then select "atom" I
- now click on each of your H's in turn, they should turn into I aoms with longer bonds
- then goto "gaussian set-up calculation"
- select "optimisation" under the "Job Type" tab
- add a descriptive title
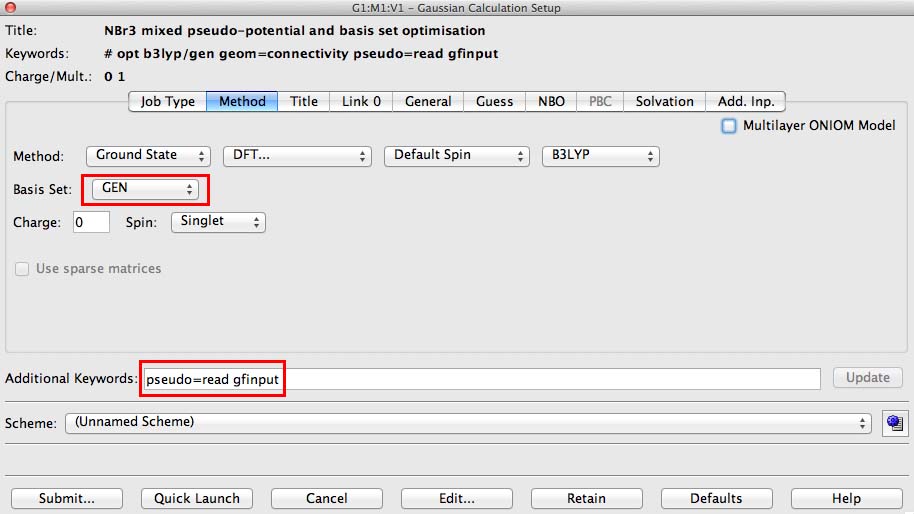
- under the methods tab on the basis button choose "GEN". This switches on the option to specify basis sets for each atom individually
- in the additional keywords section add pseudo=read gfinput. This switches on the option to specify pseudo-potentials for each atom individually and it turns on a printing option that is very useful if you make a mistake.
- then save your file, but do not submit it, ie press the submit option, save the file and then give no for the actual submission
- close the file
- now you need to add some information directly to the file (this is an advanced technique which gaussview does not support yet!)
- open the .com file you have just created
- go to the results tab and select view file, a window should open with your input file ready to edit.
- your file should look something like this:
%chk=ni3_opt_mixed.chk # opt b3lyp/gen geom=connectivity pseudo=read gfinput NI3 mixed pseudo-potential and basis set optimisation 0 1 N -0.00000000 0.00000000 0.11923374 I 0.00000000 1.86888647 -0.67333384 I -1.61850316 -0.93444324 -0.67333384 I 1.61850316 -0.93444324 -0.67333384 1 2 1.0 3 1.0 4 1.0 2 3 4
- Now we need to specify the basis sets for all atoms and the pseudo-potential for the I atoms. This is done after the coordinates and connectivity information. I have an example below for the NI3 molecule where I want a full basis set on the N but PPs on the I atoms:
%chk=ni3_opt_mixed.chk # opt b3lyp/gen geom=connectivity pseudo=read gfinput NI3 mixed pseudo-potential and basis set optimisation 0 1 N -0.00000000 0.00000000 0.11923374 I 0.00000000 1.86888647 -0.67333384 I -1.61850316 -0.93444324 -0.67333384 I 1.61850316 -0.93444324 -0.67333384 1 2 1.0 3 1.0 4 1.0 2 3 4 N 0 6-31G(d,p) **** I 0 LanL2DZ **** I 0 LanL2DZ
the general format is the following:
coordinates
(blank line)
atomic symbols (space) 0 (zero)
normal basis set
**** (four stars)
atomic symbols (space) 0 (zero)
pseudo-potential basis set
**** (four stars)
(blank line)
atomic symbols (space) 0 (zero)
pseudo-potential
(blank line) important!
- add the extra information required into your file
- save your file in the text editor, close the text editor
Run your job
- in real research we don't run jobs on our desktop, we submit them to a high performance computer. Large "queues" which many people use are set-up. Sometimes jobs can take days or even weeks to run. Yours won't!
- to give you a feeling for the research experience, you should submit this "larger" job to the Imperial cx1 hpc cluster.
- Instructions for submitting a file to the Scan servers.
- sometimes the servers are not running well, if this is the case, then you can run this job on your local computer
- if this is the case open your saved job in gaussview, select "calculate", check on the "Add.Inp" tab that your PP and basis set data is present. Then "submit" the job! You will need to add the logfile to your wiki rather than a DOI.
Check that your optimisation of NI3 has been succesful
- Visualise the final log file in gaussview to ensure it "looks" right, none of the atoms have dissociated or strange bonding patterns have emerged.
- Check the "Summary" data is everything as it should be? Check the method, the basis-set, the symmetry and the gradient.
- Confirm that your optimisation has worked opening the .log file and checking the "Item" table, does each line say "YES".
- ONLY if your job has converged properly deposit the final optimised NI3 file in the chemical database "D-space". Instructions for adding a file to DSpace and including a link on your wiki.
Update your wiki
- Link to your completed B3LYP/6-31G(d,p)LANL2DZ NI3 frequency file on your wiki
- Include the "summary" table in your wiki
- Include the "Item" table in your wiki
- Include the low frequencies lines in your wiki
- Include a Jmol image of your optimised geometry in your wiki
- what is your optimised N-I distance?