
Part C: Individual Projects
Project 1: Isomers of Mo(CO)4L2
In the second year inorganic chemistry lab course you prepare and then examine the isomers of Mo(CO)4L2 where L=PPh3 or L=piperidine (HNC5H5)by infrared spectroscopy. The number of CO vibrational bands active is related to the symmetry of the complex, four carbonyl absorption bands are expected from the compound with cis ligands and only one band is expected from the compound with trans ligands.
Your job is going to be to use calculations to predict the thermal stability and spectral characteristics of the two isomers.
Carry out all of your calculations at the HF level. Start by using a low level basis set and PP (like LANL2MB on the Mo and 3-21G on all the other atoms) to get the rough geometry right, and then for the final geometry use LANL2DZ on the Mo and 6-31G(d) on all the other atoms. To use mixed basis sets like this you will need to investigate using the "gen" keyword. To ensure you have done this correctly (before carrying out a large sequence of calculations!) get one of the demonstrators to check your files.
Key tasks are to:
- compute ground state structures cis and trans of Mo(CO)4L2 where L=PH3
- compare the geometry with computational and experimental literature values
- compute IR frequencies
- compare with YOUR experimental values (2nd year lab!)
- what is the energy difference between these isomers? Which is more stable?
if you complete the above then consider doing one or more of the following extensions
- do the same for W(CO)4L2 where L=PH3
- do the same for Cr(CO)4L2 where L=PH3
- do the same for Mo(CO)4L2 where L=NC5H5
References
ref1 "Steric contributions to the solid-state structures of bis(phosphine) derivatives of molybdenum carbonyl. X-ray structural studies of cis-Mo(CO)4[PPh3-nMen]2 (n = 0, 1, 2)", F. Albert Cotton, Donald J. Darensbourg, Simonetta Klein, and Brian W. S. Kolthammer, Inorg. Chem., 21, (1982), p294-299ref2 "Intramolecular hydrogen bonding and cation ą-interactions affecting cisĐtrans isomerization in tungsten hexacarbonyl derivatives of 2-pyridyldiphenylphosphane and triphenylphosphane", Leeni Hirsivaara, Matti Haukka and Jouni Pursiainen, Inorg. Chem. Comm., 3 (2000) p508-510
ref3 "The crystal and molecular structure of trans-tetracarbonylbis(triphenyl-phosphine)chromium(0) in a new unit cell: Is the trans conformer more stable than the cis?", Dennis W. Bennett, Tasneem A. Siddiquee, Daniel T. Haworth, ShariffĘE.ĘKabir and FarzanaĘK.ĘCamellia. J. Chem. Cryst., 34 (6) (2004) p353-359
Project 2: Boron based acids
In the first year (at Imperial anyway) the Lewis acidity of various compounds is discussed. Lewis acidity being the ability of a substance to act as an electron pair acceptor. Boron compounds of the type BX3 (we are taught) are electron deficient, the B atom contributes 3e to its valence shell and then each "X" contributes another e, giving a total of 6e, when really 8e are required to reach a full octet, and hence boron compounds readily accept additional electrons.
This being the case, where X=halide we would expect the acidity of BX3 compounds to decrease from BF3 > BCl3 > BBr3 since, this is the order of their electronegativity, F will withdraw more electron density from the B center making it even more Lewis acidic.
 However, the Lewis acidity of these compounds progresses as BBr3 > BCl3 > BF3. The explanation for this reversal of the expected trend is placed on the p-AOs of the halide, these orbitals can form a ą-like interaction with the boron atom, where the lone pairs on the halide back-donate electron density into the empty pAO on the boron.
However, the Lewis acidity of these compounds progresses as BBr3 > BCl3 > BF3. The explanation for this reversal of the expected trend is placed on the p-AOs of the halide, these orbitals can form a ą-like interaction with the boron atom, where the lone pairs on the halide back-donate electron density into the empty pAO on the boron.
 However, even this hypothesis has been downgraded and molecular orbital arguments have been used to discuss the relative acidity of boron based acids. BCl3 is inherently a stronger acid than BF3, it forms a stronger B-X bond and a more stable adduct with NH3. This is in part due to the LUMO of BCl3 lying closer in energy to the HOMO of NH3 resulting in a stronger interaction between these orbitals.
However, even this hypothesis has been downgraded and molecular orbital arguments have been used to discuss the relative acidity of boron based acids. BCl3 is inherently a stronger acid than BF3, it forms a stronger B-X bond and a more stable adduct with NH3. This is in part due to the LUMO of BCl3 lying closer in energy to the HOMO of NH3 resulting in a stronger interaction between these orbitals.
In this project your job is to investigate the boron based acids BF3 and BCl3 with the lewis bases NH3, NF3 and NCl3.
Key tasks are to:
- optimize structures individually
- optimize structures in the adducts
- look at the HOMO and LUMO orbitals
- look at the charge on the central B and N atoms
if you complete all of the above then do the same for B(OH)3, B(NH2)3, and B(CH3)3
Reference
ref1 "Why is BCl3 a stronger Lewis acid with respect to strong bases than BF3?", F. Bessac and G. Frenking, Inorg. Chem., 42 (2003), p7990-7994
Project 3: Gold interactions with water
Pure gold is not very reactive, however nano-structured gold supported on an oxide surface is catalytically active. Water is often present and its interaction with gold clusters and surfaces is not well characterised. Part of this project is to look at how water interacts with a gold dimer.Gold also exhibits particularly large relativistic effects. The electrons close to the gold core are moving near the speed of light and this causes a relativistic mass increase in the electron and hence a contraction in the radial extent of some orbitals. s orbitals are affected most because the s electrons spend more time near the core. In addition each orbital has to be orthogonal to all the other orbitals. These effects combine in Au to make the relativistic contraction of the 6s orbital very large, which in-turn pushes out the p and d shells as these are now better shielded from the core. These effects play an important role in the chemistry of gold, and can be examined by using "relativistic" and "non-relativistic" pseudo potentials on gold.
In this project your job is to investigate water interacting with "relativistic" and "non-relativistic" small gold clusters.
Carry out all of your calculations at the HF level. Lower level pseudo-potentials are inaccurate, so you will have to start directly with the "higher level" basis sets. Use the MHF60 and MDF60 pseudo-potentials and a 6-31G(d,p) basis set on the water atoms. To use mixed basis sets like this you will need to investigate using the "gen" keyword. To ensure you have done this correctly (before carrying out a large sequence of calculations!) get one of the demonstrators to check your files.
- optimize the relativistic and non-relativistic geometry of Au2
- compare the relativistic and non-relativistic geometries of Au2
- compare your geometries to literature values
- optimize the geometry of H2O
- optimize a number of possible modes of interaction for Au2 and H2O
- calculate the vibrational spectrum, what happens to the O-H modes?
- compare the relativistic and non-relativistic geometries and dissociation energies
- which is the most stable conformer?
here are some possible interaction modes:
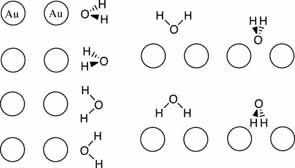
if you complete all of the above then consider doing one or more of the following extensions:
- look at the relativistic and non-relativistic MOs of Au2
- look at the interaction of Au2 with NH3
- look at the interaction of Au2 with SH2
Reference
ref1 "Relativistic effects in gold chemistry. I. Diatomic gold compounds", Peter Schwerdtfeger, Michael Dolg, W. H. Eugen Schwarz, Graham A. Bowmaker, and Peter D. W. Boyd, J. Chem. Phys, 91 (1989), p1792ref2 "Relativistic coupled cluster calculations for neutral and singly charged Au3 clusters", Ralf Wesendrup, Tricia Hunt, and Peter Schwerdtfeger, J. Chem. Phys, 112 (2000), p9356