
Molecular Orbitals in Inorganic Chemistry
Transition metal MO diagrams
Q:I'm confused the rules seemed to have changed between building a main group MO diagram and a TM MO diagram?
A:You are correct the rules have changed! It is important to realise that we treat the TM-MO diagram DIFFERENTLY from main group MO diagrams. We are no-longer building a MO diagram from scratch, TM MO diagrams ALWAYS start from the octahedral case. All changes are now considered as SMALL perturbations to this diagram! (for this course!!)What we need to remember is that the MO diagrams we build include Δε of the orbitals and overlap (S) what they don’t include are the
quantum mechanics
of exchange and correlation, which is a very non-intuitive quantity.TM especially the 3d exhibit strong exchange/correlation effects because the dAOs are very contracted and the electrons in these orbitals can
see each other
and want to avoid interactions. These strong exchange/correlation effects make it very hard to make an educated guess based simply on easy parameters like Δε and overlap.So with TM we start with the octahedral diagram as I have given it to you. How do I know it looks like this? Because when we calculate the MOs this is how they come out! The basic Oh diagram is the result of the real QM with the exchange/correlation components included.
Q:Why does delta octahedral increase down a group for TMs?
A:This is a surprisingly common question especially as regards the organometallics and TM chemistry course. As with much of real chemistry as you dive in the answers get more and more complex. First of all there is a contrast with main group, main group bonds get weaker down a group. A typical explanation is that the orbitals are more diffuse and overlap is worse. Now for the TMs, the bonds get stronger down a group! Why is this? There are no inner d orbital angular momentum shells and thus the 3d orbitals are highly contracted (see the next question!) this means the 3d electrons shield the nucleus quite well and that they are concentrated (lots of e-e correlation and repulsion). However, this contraction also means the 3d AOs have a comparatively very poor spatial overlap with ligand orbitals, the two cannot get close enough to overlap well. The net result is weak metal ligand interactions and a weak delta octahedral splitting. Going down the group to the 4d and 5d orbitals, these are nicely shielded by the 3d orbital shell, they extend further into space, and there is a much better overlap with the ligand orbitals, combined with a slight reduction in density, overall the balance is such that bonding gets stronger and there is a larger delta octahedral splitting. Qualitatively, in terms of the energy level diagram the energy gap between the valence dAOs and ligand FOs is reduced leading to a stronger interaction.
Q:Why do the metal 4s AO and a1g ligand FOs interact, are they not too far apart in energy? Similarly why do the 4p AOs and the t1u ligand FOs interact?
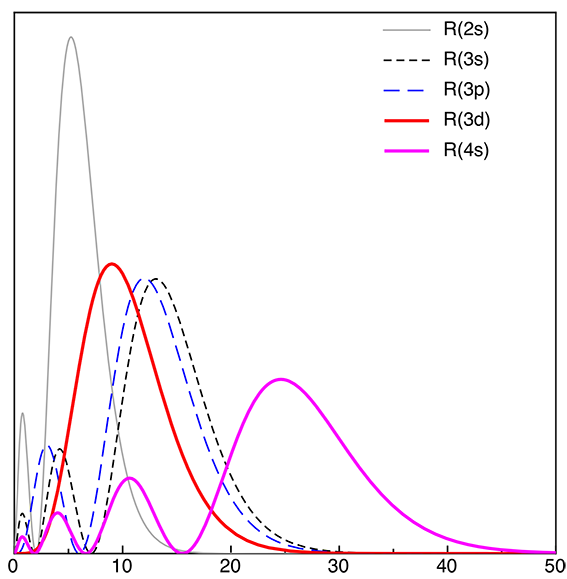
A:First remember that TM complexes break the rules. So all the nice tidy rules we had for main-group elements do not hold as happily for TMs. Then remember that this diagram is simply what comes out of the fundamental QM! (see the answer to Q1)Having established that the QM provides our basic reference system, there are some additional "conceptual" hints which support this result. You should remember from foundation that the 3d and 4s orbitals are actually very close in energy (go back and look!) so the basic Oh a actually a very "expanded" diagram compared to the main group complexes we have studied up to now.
Also, take a look at the radial extent of the 3d vs the 4s AOs plotted above (the 4p are close to the 4s), look how far out the 4s AOs are, they will overlap much better with the ligand FOs than the contracted 3d AOs.
Actually the situation is not so clear cut, the 4s AOs have disparate energy (relative to the ligand FOs) but good overlap spatially (perhaps not so good when we take into account that these orbitals are more diffuse, ie less dense) while the 3d AOs have close energies but very poor overlap (but are also more dense).
It would be nice if the physical world was simple and could be described by simple rules which are "the truth", but this is not the case and we have to try and make sense of this complexity as best we can. So do not forget that the "rules" are just us trying to organise and make order out of a complex system and they are not "the truth". The important point is to understand this complexity.
Q:Why is the ndAO and (n+1)s and (n+1)p AO splitting for M2 well separated in the notes of L5 while in the answers for the self-study problem on Mo2 and W2 the d-AO splitting is greater and overlaps with the s than p-AO splitting?
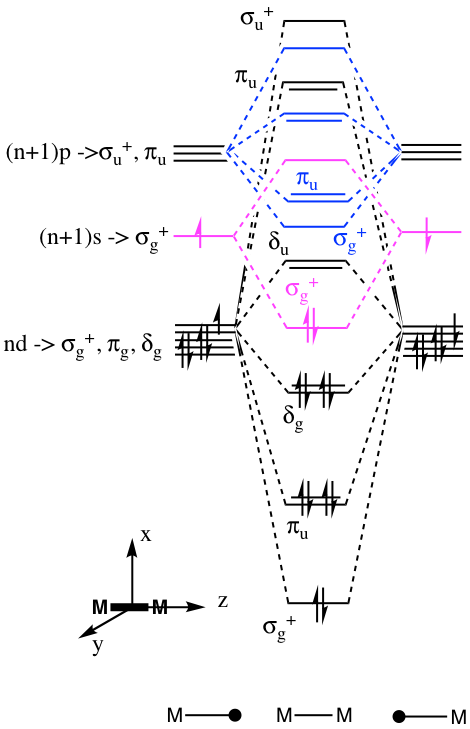
A:When introducing new material I have tried to simplify what is going on to help you gain understanding in a graduated manner. In the notes I split the various levels into clearly separate sections and I am also consistent with the “main group” rules for the size of the splitting. However, in reality (as is so often the case with TMs!) life is more complex. In reality the energies of the nd and (n+1)s/(n+1)p AOs can overlap, particularly if n>3 (i.e. for beyond the 3d TMs). 4d and 5d TM have orbitals that overlap better and split more than 3d metals (this is some of the reason behind why bonding gets stronger down a group for TMs). The answer in the self-study problem introduces some of this complexity. We would need to carry out a calculation to know exactly the correct order for the orbitals. However, we can make an educated guess knowing that Mo and W are 4d and 5d, and that the (n+1)s gap with respect to the nd gets smaller down a group. I have pushed the antibonding MOs for the dAOs up a long way, they may not go quite that far, it is hard to estimate these.
Q:I have a problem concerning homonuclear diatomic transition metal MO diagrams. In the tutorial question, it asks us to draw a MO diagram for diMolybdenum. From our notes, it shows the s-orbitals to be far in energy from the d-AOs and so the lowest lying bonding orbital from the s-AO overlap is higher in energy than the highest energy antibonding orbital of the d-AO overlap. Therefore, I was wondering what makes the s-AO shift down in energy in diMolybdenum? Would it be due to a similar explanation as the s-p gap increasing across the PT and so the s-d gap increases too?
A: In the notes I wanted to start with a simple example, and so I started with a TM where the nd-(n+1)s gap is large. The real situation is quite complex! The energy of an orbital depends on its occupation, thus if the occupation changes the orbital energy changes. For a given configuration the nd subshell is stablised relative to the (n+1)s subshell across the series. However, the difference in the electron-electron repulsion and exchange energies for a 3dn-14s1 vs a 3dn4s2 configuration means that the energy "difference" between the two configurations is not a simple difference in energy between the frozen 3d and 4s orbital energies. The difference in energy taking into account electron electron exchange and repulsion shows a decrease in the "sd gap" across the series. In addition there is the "half filled shell" stability (maximum stabilisation of the exchange energy) for Cr and Cu where a 3dn4s2 with n=5 and 10 respectively, configuration is favoured. In this question I am expecting you to recognise that the (n+1)s (n=3,4 and 5 respectively) for Cr, Mo and W are doubly occupied and thus to "guess" that (n+1)s orbitals are potentially "stabilised" for these atoms. Of course, in reality it is not just the orbitals that are stabilised but the configurations, which lead to orbital stabilisation. For those looking for a fuller explaination here is an article: Why the 4s orbital is occupied before the 3d by M.P. Melrose and E.R. Scerri, J. Chem. Ed., 1996, 73, p498 http://pubs.acs.org/doi/pdf/10.1021/ed073p498
Q:I'm confused about the two descriptions of the ligand orbitals, L5 Figure 11 and L5 Figure 13?
A:the first thing to realise is that we treat the TM-MO diagram DIFFERENTLY from main group MO diagrams. We only include certain information, it is assumed you are aware of the rest. When constructing a TM-energy diagram to show all the ligand orbitals is too much work, so we only show those that interact with the metal are still there we just assume you know this and then ignore them!) So in this case the only orbital we are interested in is the one with a lobe pointing towards the metal, like that below for EH3.
The second thing to realise is that small perturbations to an orbital do not change the overall symmetry of the orbitals dramatically. Yes the symmetry is formally "broken" but in actual fact there is not a large effect on the MOs. So we can make the simplifications shown above. The "sp-like" FO are simplifications of the real FOs involved, for example in the case of NH3 or PH3 or SiH3 or CH3. The first representation below is using the H 1sAOs and the second is using ligand L "sp-like" FOs.
Q:Why are the orbitals in L6 Figure 19 and 20 split?
A:These figures refer to the reduction in symmetry from Oh to C4v for the metal and ligand fragment orbitals respectively. As the point group changes, the way the orbitals respond to the symmetry elements stays the same. However, now there are less symmetry elements (look at the top row of C4v compared to Oh). Thus the way we label the symmetry of the orbitals has changed.
Nevertheless, the metal orbitals are still degenerate. Perhaps you missed it in the lecture when I said that I separated the energy lines to help you identify the labeling you should expect? As this has been a common question I've created an updated diagram that better represents the situation, see the first figure below.
The ligand orbitals which do not contain the new apical ligand fragment remain the same energy, however those that contain the new frament will change in energy slightly. The change will depend on the exact ligand and so will the energy ordering of the levels. See the second figure below, here I have used a slightly larger new component which increases the bonding and antibonding interactions slightly.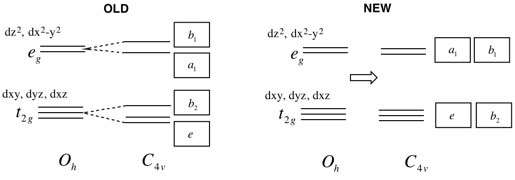
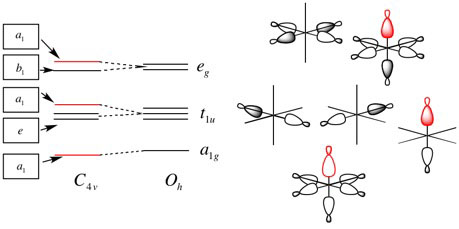
Q:I'm confused, in L6 we derived first the Oh energy diagram and then the C4v energy diagram by changing the apical sigma ligand, as shown below. Why is the 3d a1 FO not interacting with the lowest energy ligand a1 FO?
A: We always start with the Oh diagram and consider changes as small perturbations Notice how the "same" energy levels interact even though the symmetry labels have changed! For example, the 3d a1 FO is not interacting with the lowest energy ligand a1 FO, it is still interacting with the same energy level as when it was an "eg" label. The "eg" level is split slightly due to the change in symmetry, but the overall the same levels interact. This is the new rule in action for TM-MO diagrams.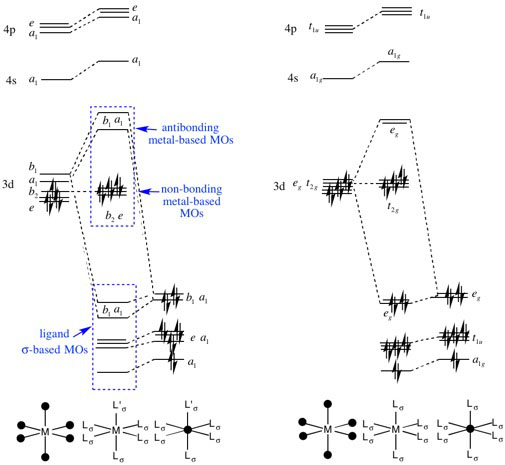
Q:In the C4v MO diagram of a TM complex, why do the e FOs of the TM remain non-bonding when they have the same symmetry label and are close in energy to the ligand e FOs?
A: The answer to this question is similar to the one above. The b2 and e orbitals come from the old Oh t2g set, these orbitals did not interact with the ligands in the Oh diagram and so they remain essentially non-interacting in the slightly perturbed case of the C4v MO diagram. A supporting hint also comes from the answer to question 1 above, the 3dAOs are contracted and so do not really extend out in space far enough to interact with the ligand sigma type orbitals, while the 4p AOs are able to interact and so they dominate.