
Molecular Orbitals in Inorganic Chemistry
Transition metal MO diagrams
Q:I was trying to go over the symmetry elements for the octahedral point group and couldn't find all the 6C2 axes
A: this one is really hard to draw, the best way I have found is to work it out "by symmetry". First, we know there is a C2 axis between each set of bonds. In the equatorial plane this gives us two C2 axes. Then for each of the axial planes (these are defined along the bonds of the equatorial ligands) there are another two C2. As there are two axial planes of this type we count four C2 in the axial planes. Combining these results we have six C2 axes in total.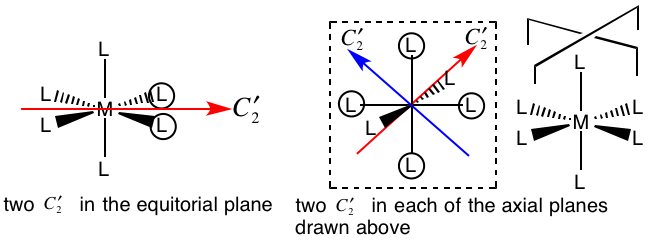
Q:Looking at the octahedral point group, the labeling of the mirror planes is confusing, one is annotated as sigma-d however I was under the impression this was sigma-h as the mirror plane depicted does not pass between bonds but instead is perpendicular to the principal C4?
A: First of all, this is a tricky one, well spotted! The problem is that we have multiple principle axis perpendicular to each other. So the same mirror plane is "σd" (or σv) for one axis and "σh" for another. So we select each C4 axis, label the mirror plane perpendicular to that axis as "σh", notice how this goes through the center of a face (red mirror plane in (b)). Then what mirror planes are left? Those that go through the edges of the cube (red mirror plane in (a)) these we call σd ("d" because they go between C2 axes).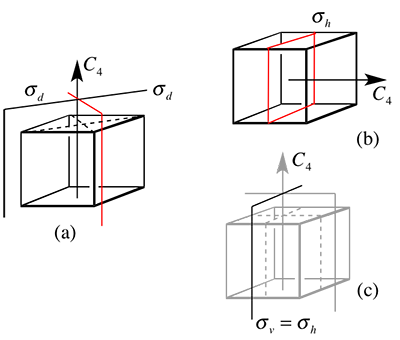
Q: In TM MO diagrams are a1 and b1 meant to be degenerate?
A: When the symmetry of a TM complex is Oh these orbitals form a degenerate e symmetry set, however once the symmetry drops the a1 and b1 (C4v) or a1g and b1g (D4h) are no longer formally degenerate. Typically the symmetry had dropped because we have different and/or new types of ligand, this means the orbital energies are also slightly different. For example, axial X(pi) ligand sigma orbitals are slightly different from the L(sigma) ligand orbitals in ML4X2.
Q:In your MO diagram of the ML4 square planar molecule (tutorial question L7) the 3a1g and 2b1g anti-bonding orbitals are raised above the 4s level of the TM. I am wondering why this is the case, as in octahedral molecule, the splitting energy is not so large, and the anti-bonding orbitals still lie below the 4s?
A: In this case you need to bring in some knowledge from your first year foundation and coordination chemistry courses. The TMs that form square planar complexes are Ni, Pd and Pt. In the electronic filling of the orbitals the (n+1)s is typically considered to be very close in energy to the nd AOs. For example, look at the electronic structure of Ni (and Pd and Pt) from web-elements. Thus, we start with a fragment orbital pattern assuming that the (n+1)s level is very low for these TMs (adding an annotation explaining this in your MO diagram would be a great idea). When I draw the "standard" ML6 diagram we assume the (n+1)s lies sufficiently above the nd that interactions are minor.The above description is substantially "simplified", and the actual situation is far more complex, see the discussion in Q2 above. We will be addressing configurations in my Spring term course.
Q: In the handout, you say that pi donor ligands have no pi* antibonding orbitals so the metal is forced to interact with the pi fragment orbital of the pi donor ligand, which lies just underneath the d AO. But in the problems class you made an MO diagram of ML4X2 where X are pi donor ligands. In the final MO diagram you've included pi and pi* fragment orbitals!!! And you've interacted the pi* FO! This is just confusing!
A: Here there is some confusion about two different types of orbital, both have pi symmetry but they are different orbitals. Remember pi is a symmetry label it is not a particular type of orbital. So when there is one pi donor there is only a single orbital, the pAO on the pi donor thus there is only one orbital to interact (no pi* FO). BUT when we have two pi donors, the two pi-dornor pAOs can interact to produce two sets of pi orbitals (now we do have a pi* set). The pi* anti bonding set are not the same as the pi* orbital on a single ligand but are the antibonding combination from two different ligands. I have attached a diagram which I hope makes this much clearer.
Q: In lecture 8 the diagram shows that the π* (pi star) orbital is above the 3d orbitals of the metal however, this is the opposite for the example you did in the problem class - why are they not of the same energy?
A: The difference is the type and number of ligands. In the problems class we looked at a pi-donor ligand, in L8 we treated a pi-acceptor ligand. Also be very careful, there is a difference between treating a single ligand with two pi orbitals, and two ligands each with one pi orbital!
Q:Could you help explain to me why the antibonding a1g orbitals in the digram from page 2 (shown below) of the L9 hand drawn MO diagram are higher in energy than the b1g orbital?
A: To answer this question you want to think about (a) where else have you seen these orbitals and (b) what is their comparative orbital character?First consider (a), you have seen these orbitals in the ML6 octahedral sigma framework, where they are degenerate. So a good first approximation would be to guess these are very close in energy. We also know that the pi-donor sigma orbitals can be a little higher in energy than the sigma-donor orbitals thus having a slightly reduced energy difference and hence slightly smaller energy gap leading to a slightly stronger interaction and slightly larger splitting energy. Comparatively this means the X2 contribution will be slightly larger leading to a greater antibonding interaction.
Now lets consider (b), there are 2 long range (through 2 bonds) bonding interactions in the b1g and 4 medium range (through space) antibonding interactions. For the a1g there are 3 long range (through 2 bonds) bonding interactions, and 4 medium range (through space) bonding interactions. There are also 8 medium range (through space) antibonding interactions, which are slightly larger due to the larger X2 contribution. If we recognise that the overlap is highly distance dependent the medium range interactions will dominate. Thus we are comparing 4 medium range (through space) antibonding interactions of the b1g with the 8 antibonding and 4 bonding medium range (through space) interactions. Thus, if these were to cancel the a1g also has net 4 antibonding medium range (through space) interactions. If we also recognise the slightly larger X2 contribution the a1g should be slightly higher in energy.
However, as these orbitals are very close in energy subtle effects may come into play, such as the Hij term which we cannot “guess” so easily. Thus, these energy levels should be close together, but I would accept the a1g lying slightly below the b1g as well, we would need to carry out a calculation for a specific ligand and metal system to determine the exact ordering. In-fact this is essentially stated on the next page of the notes in the large MO diagram (regarding MOs C and D).
Q: I have a question about one of the exam questions. As illustrated below, the first question (i) was to draw the 4e and 4a1 orbitals. The 4e1 orbitals seem logical, and so does the 4a1 orbital, until I read in the answer key, which stated that the metal contribution to the 4a1 orbital was bigger than the ligand contribution. In normal circumstances I would follow the same reasoning, but due to the presence of the pi acceptor ligand (which is slightly higher in energy than the metal d orbitals), I was unsure as to why the metal contribution was higher to the MO than the ligand contribution.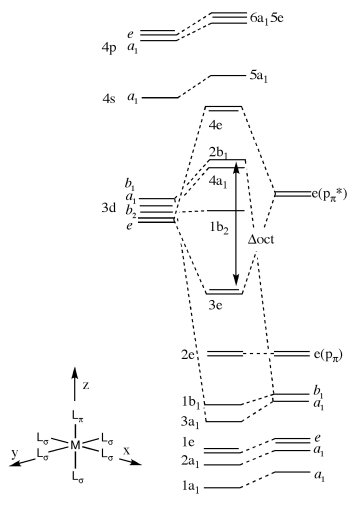
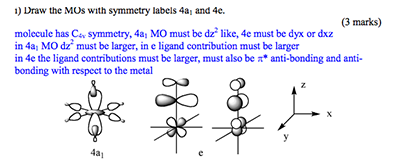
A: Careful, you need to look very carefully at the diagram, it tells you what to do and which ligand orbitals are involved. The 4a1 is an antibonding MO formed with the a1 ligand **sigma framework** and the dAOs, as this orbital is antibonding the higher lying fragment is the largest contributior and in this case it is the dAO. The sigma orbitals are almost always deeper than the dAOs, it is only the pi orbitals that come close to the dAOs in energy. The 4e MO is overall antibonding and so the highest energy FO contributes most, in this case it is the p(pi)antibonding FO of the ligand which is higher in energy than the dAOs.
Q:I am getting a little bit confused as to labelling the delta octahedral splitting parameter. In lecture 7 you discuss two delta Oct; one for the pi-donor ligand and one for the complex.A: The problem is that delta Oct is an old parameter, based on experimental data, on a model ML6 complex (ie identical ligands) and formed in the days of CFT and so it does not relate especially well to modern MO theory. Lets start with all sigma ligands ML6, L sigma-donor delta Oct is easily defined between the t2g and eg MOs (not shown on the figure below). In a UV-vis spectrum we might (to a first guess) expect one peak (actually things are more sophisticated that this and we cover this aspect next year in Advanced Spectroscopy)
Then we can make one ligand different, in this case the symmetry point group drops to C4v and the upper eg level splits slightly, see the far left hand side of the figure below. The old t2g levels remain degenerate but get new symmetry labels, b2 and e. We might expect to see one very broad peak in the UV-vis covering the two very close energy levels. Sigma-donor delta Oct is still the maximum spread of the dAOs (shown in green).
Now consider the far right hand side of the figure below, the case of six pi-donor ligands. We have octahedral symmetry and so the old t2g and eg labels are back, and pi-donor delta Oct is well defined (shown in pink). In a very simplified model we might expect to see a single peak in the UV-vis spectrum
Now for the complex bit! Lets consider a complex with one pi-donor ligand and the rest are sigma donors, this is the middle diagram in the figure below. In essence we can think about two delta Oct parameters, one is the sigma-donor delta Oct (blue) and one is the pi-donor delta Oct (red). In the UV-vis spectrum we might expect to see two peaks, one for each "type" of excitation.
What then is the delta Oct for the whole molecule? In reality this is hard to define, we might want to say it is the largest spread of the dAOs, ie the ligand field splitting energy, the amount by which the ligands split the dAOs, and so in this case would it be the same as the sigma-donor delta Oct.

Q: I have question regarding strong and weak field ligands. At the end of Lecture 7 we classified CN- as a strong field ligand and this was due to its pi* acceptor properties pushing the e molecular orbitals down and increasing delta octahedral. However, in Table 1 of lecture 8 it is then pointed out that the CN- ligand only has very weak pi-acceptance. I am confused!A:Using spectra (ie the spectrochemical serires) and hence relating to delta octahedral CN- is classed as a strong field ligand, moreover because CN- is isoelectronic to CO this makes some sense. Thus based on the empirical information to hand, this was not a bad conclusion to make. So historically (and even still now!) CN- is classed as a stong pi acceptor ligand.
More recently computational methods have reached the point where we can compute transition metal complexes, and we are finding that some of these older ideas are not consistent with what QM tells us. For example as in the population analysis data in Table 1 from lecture 8. Note the paper is from 2001 so almost 20 years ago and this new understanding has still not percolated into mainstream chemistry. People are very resistant to change, especially when something is more complex/complicated when compared to a nice easy to understand simple idea.
So this is were you have to hold the “old ideas” in your head, knowing they are not quite right but that most people without your knowledge will be thinking this way, AND you have to keep track of the “new ideas” which more accurately represent reality. We really need to go back and look at many of the older ideas again and develop new rationalisations/models of understanding.
So, if something as simple and core as these ideas are wrong what else are you being taught that is not quite correct?? Probably quite a bit, so it is always best to keep an open mind!